

International Journal of Cancer and Clinical Research
A Derivative of Differentiation-Inducing Factor-3 Inhibits PAK1 Activity and Breast Cancer Cell Proliferation
Peter Oladimeji1, Yuzuru Kubohara2,3, Haruhisa Kikuchi4, Yoshiteru Oshima4, Courtney Rusch1, Rebekah Skerl1, Maria Diakonova1*
1The Department of Biological Sciences, University of Toledo, Ohio, USA
2Institute for Molecular & Cellular Regulation, Gunma University, Maebashi, Japan
3Graduate School of Health and Sports Science, Juntendo University, Chiba, Japan
4Graduate School of Pharmaceutical Sciences, Tohoku University, Sendai, Japan
*Corresponding author:
Maria Diakonova, The Department of Biological Sciences, University of Toledo, 2801 W. Bancroft Street, Toledo, Ohio, USA, Tel: 419-530-7876, Fax: 419-530-7737, E-mail: mdiakon@utnet.utoledo.edu
Int J Cancer Clin Res, IJCCR-2-023, (Volume 2, Issue 4), Original Article; ISSN: 2378-3419
Received: July 23, 2015 | Accepted: September 15, 2015 | Published: September 17, 2015
Citation: Oladimeji P, Kubohara Y, Kikuchi H, Oshima Y, Rusch C, et al. (2015) A Derivative of Differentiation-Inducing Factor-3 Inhibits PAK1 Activity and Breast Cancer Cell Proliferation. Int J Cancer Clin Res 2:023. 10.23937/2378-3419/2/4/1023
Copyright: © 2015 Oladimeji P, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Differentiation-inducing factors 1-3 (DIFs 1-3), chlorinated alkylphenones identified in the cellular slime mold Dictyostelium discoideum, are considered anti-tumor agents because they inhibit proliferation of a variety of mammalian tumor cells in vitro. Although the anti-proliferative effects of DIF-1 and DIF-3 are well-documented, the precise molecular mechanisms underlying the actions of DIFs have not been fully elucidated. In this study, we examined the effects of DIFs and their derivatives on PAK1, a key serine-threonine kinase, which is activated by multiple ligands and regulates cell proliferation. We examined the effect of DIF derivatives on PAK1 kinase activity in cells. We also examined the effect of DIF-3(+1) derivative on PAK1 kinase activity in vitro, cyclin D1 promoter activity and breast cancer cell proliferation. It was found that some derivatives strongly inhibited PAK1 kinase activity in human breast cancer MCF-7 cells stably over expressing PAK1. Among the derivatives, DIF-3(+1) was most potent, which directly inhibited kinase activity of recombinant purified PAK1 in an in vitro kinase assay. Furthermore, DIF-3(+1) strongly inhibited both cyclin D1 promoter activity and proliferation of MCF-7 and T47D breast cancer cells stably over expressing PAK1 in response to prolactin, estrogen, epidermal growth factor and heregulin. In the present study we propose PAK1 as DIF-3(+1) target mediating its anti-proliferative effect.
Keywords
Differentiation-inducing factor-3, PAK1 kinase, Cell proliferation, Breast cancer, Cyclin D
Abbreviations
Ab: Antibody, DIF: Differentiation-inducing Factor, JAK2: Janus kinase 2, PAK1: p21- activated kinase 1, PRL: Prolactin, E2: 17-β estradiol, EGF: Epidermal Growth Factor, HRG: Heregulin, vcl: vehicle, WT: Wild Type
Introduction
Differentiation-inducing factors 1-3 (DIFs 1-3) are chlorinated bioactive compounds identified in the slime mold Dictyostelium discoideum, which induce stalk cell differentiation [1-3]. Among them, DIF-1 is the most active in inducing stalk-cell differentiation, while DIF-2 has less activity than DIF-1. DIF-3, which is the first metabolite formed during DIF-1 degradation, has the lowest activity to induce stalk cells differentiation [3-5].
DIFs are considered anti-tumor agents because they inhibit proliferation of mammalian tumor cells in vitro, including leukemia [5-9], cervical [10], gastric [11], and colon [12] tumor cells. Activity of different DIFs to inhibit proliferation of mammalian tumor cells is converse to the activity demonstrated for differentiation of D. discoideum stalk cells; the most potent anti-tumor agent is DIF-3 [5,7,10]. Moreover, DIF-1and DIF-3 inhibit tumor growth in mice in vivo [12,13].
Different intracellular pathways have been implicated in anti-proliferative effect of DIFs. Thus, DIF-3 reduces the expression of cyclin D1 mRNA by inhibiting Wnt/β- catenin signaling [12] and that DIF-1 and DIF-3 reduce protein level of cyclin D1 by accelerating its degradation via activation of GSK-3β [10,12,13]. In addition, DIF-1 inhibits proliferation of leukemia cells through inhibition of ERK and STAT3 signaling [8,11]. It has also been shown that DIFs are direct inhibitors for PDE1 (calmodulin-dependent phosphodiesterase) [9].
Taken together, these findings shed a light on possible mechanisms of anti- proliferative effect of DIFs on tumor cells. However, the precise molecular machinery underlying the anti-proliferative action of DIFs remains to be elucidated.
PAK1 is a serine/ threonine kinase which regulates different intracellular functions including proliferation, cytoskeletal dynamics, cell survival, and gene transcription (reviewed in [14]). Altered expression and/or activation of PAK1 is evident in various cancers (reviewed in [15,16]). The role for PAK1 in breast cancer has been studied to the most extent (reviewed in [14,15]). The PAK1 gene amplifications are found in 17% of breast cancer [17,18]. Over expression of PAK1 in human breast tumors correlates with tumor grade [17-22]. In a transgenic mouse model, PAK1 hyper activation leads to mammary gland tumors [23]. PAK1 plays a critical role in premalignant progression of MCF10 series of human breast epithelial cell lines [24]. PAK1 activation has been linked to ErbB2-dependent transformation of mammary epithelial cells [25,26]. Recently, our lab has demonstrated a role for prolactin (PRL)- mediated tyrosyl phosphorylation of PAK1 in breast cancer cell motility, adhesion and invasion (reviewed in [27]). Thus, PAK1 has become one of the focal points in the investigation into the mechanism and onset of human breast cancer, and PAK1 inhibition represents plausible drug target in breast cancer.
In this study, to further corroborate the potential of DIFs as anti-proliferative agents, we investigate the effects of various DIF derivatives on PAK1 kinase activity. We show here that the different DIF derivatives inhibit PAK1 activity to varying degrees with the strongest inhibition in cells treated with DIF-3(+1) derivative. More importantly, it was found that the DIF-3(+1) directly inhibits PAK1 kinase activity in vitro. We also show here that DIF-3(+1) inhibits both cyclin D1 promoter activity and proliferation of MCF-7 and T47D breast cancer cells stably overexpressing PAK1. These results suggest that PAK1 is a direct target molecule of DIF-3(+1) which mediates the anti-proliferative effect of DIF-3(+1) on breast cancer cells.
Materials and Methods
Cell cultures
Parental and MCF-7 cells stably expressing HA-tagged PAK1 WT [28] were maintained in DMEM (Corning Cellgro) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich). Parental and T47D cells stably expressing myc-tagged PAK1 WT [29] were maintained in RPMI 1640 medium (Corning Cellgro) supplemented with 10% FBS.
Reagents and antibodies
DIF derivatives (Figure 1) were synthesized as described previously [5]. Human PRL was purchased from Dr. Parlow (National Hormone and Peptide Program NIDDK). Heregulin (HRG), epidermal growth factor (EGF) and estrogen (E2) were purchased from ProSci Inc, BD Biosciences and Sigma-Aldrich, respectively. Polyclonal αPAK (N- 20; Santa Cruz Biotechnology), and monoclonal αHA (Covance) antibodies were used in this study.
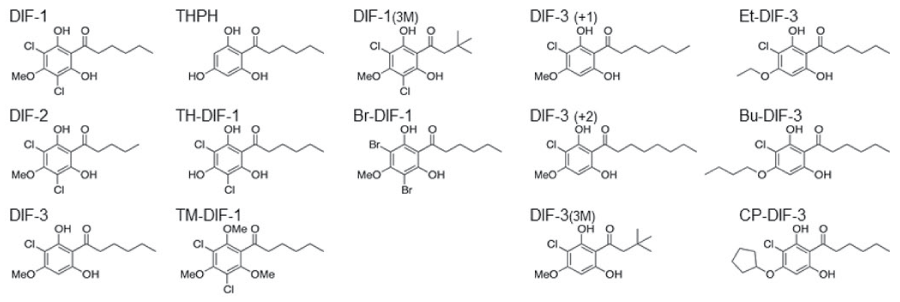
.
Figure 1: Chemical structure of DIF derivatives.
DIF derivatives were synthesized as described previously in Gokan et al. 2005 [5].
View Figure 1
in vitro kinase assay
To assess PAK1 kinase activity, HA-PAK1 was immunoprecipitated with anti- HA antibody from cells treated with either vehicle, PRL (200 ng/ml, 20 min, E2 (1 nM, 30 min), or HRG (30 ng/ml, 15min) and DIF derivatives (20 μM for the indicated time) and subjected to an in vitro kinase assay in kinase buffer (50 mM HEPES, 100 mM NaCl, 5 mM MnCl2, 0.5 mM dithiothreitol, 1 mM Na3VO4; pH 7.6) in the presence of 10 μCi [γ- 32P] ATP (MP Biomedical), and 5 μg of histone H4 (substrate of PAK1; New England Biolabs) 10 μg/ml aprotinin, and 10 μg/ml leupeptin at 30℃ for 30 min. Relative levels of incorporation of 32P into histone H4, an indicator of phosphorylation, were assessed by autoradiography and estimated by a phosphoimager screen. The same membrane was blotted with αHA to assess the amount of PAK1 for each condition. PVDF patterns were scanned and the amount of PAK1 was quantified using Multi-Analyst (Bio-Rad Laboratories) software. Relative PAK1 kinase activity was then normalized by the amount of immunoprecipitated PAK1 for each lane.
To assess direct inhibition of PAK1 by DIF-3(+1), GST- tagged PAK1 were purified using a glutathione-agarose affinity columns (Sigma-Aldrich). The purity of the proteins was monitored by SDS-PAGE. GST-tagged PAK1 was incubated with DIF- 3(+1) or THPH at 30℃ for 30 min in kinase buffer containing 10 μCi of [γ- 32P] ATP, 10 μg/ml aprotinin, and 10 μg/ml leupeptin. Proteins were separated by SDS-PAGE and visualized by autoradiography followed by immunoblotting with indicated Ab.
Luciferase reporter gene assay
Luciferase assay was performed as described previously [30]. MCF-7 cells stably over expressing PAK1 WT were co-transfected with cyclin D1-luciferase reporter and pCH110 plasmid containing a functional lacZ gene. The cells were serum deprived for 24 h, treated with either vehicle, 200 ng/ml of PRL, 1nM E2, 30 ng/ml HRG or 100 ng/ml EGF with or without 5 μM DIF-3(+1) for an additional 24 h. The cells were lysed, and luciferase activity was measured using Luciferase assay kit (Promega) according to the manufacturer's protocol. Luciferase values were corrected for transfection efficiency by determining the ratio of luciferase activity to β-galactosidase activity and expressed as "normalized cyclin D1-luciferase activity." Each transfection was performed in triplicate wells.
Cell proliferation assay
MCF-7 and T47D cells stably expressing PAK1 WT (5 × 103 cells/well) were serum-deprived and allowed to grow for 7 days in 96-well plates. Each well contained 200 μl of DMEM (for MCF-7 clone) or RPMI (for T47D clone) were treated with either vehicle, 500 ng/ml of PRL, 1nM E2, 30 ng/ml HRG or 10 ng/ml EGF with or without 5 μM DIF-3(+1). In some experiments 5 μM IPA-3 (Tocris) was added. Cell proliferation was assessed by MTT cell proliferation assay (Life Technologies) according to manufacturer's protocol.
Statistical analysis
Data from at least three separate experiments per each condition were pooled and analyzed using one-way ANOVA plus Tukey's HSD test. Differences were considered to be statistically significant at P < 0.05. Results are expressed as the mean ± standard error (S.E.).
Results and Discussion
DIF-3 and its derivatives inhibit PAK1 kinase activity in cells
We have previously demonstrated that PRL-activated JAK2 tyrosine kinase phosphorylates and activates PAK1, resulting in the activation of cyclin D1 promoter activity and invasion of human breast cancer cells [30-32]. We thus assessed an effect of DIF-3 on PAK1 kinase activity because DIF-3 is the most potent anti-tumor agent among the original DIFs [5,7,10]. We treated MCF-7 cells stably expressing HA-PAK1 with DIF-3 over a time course followed by PRL treatment for 20 min. Immunoprecipitated PAK1 was assessed for PAK1 kinase activity by an in vitro kinase assay with histone H4 as a substrate. As expected, PRL increased PAK1 kinase activity by 2.6-fold, which was inhibited by DIF-3 in a time-dependent manner and reached the basal level by 2 h (Figure 2A).
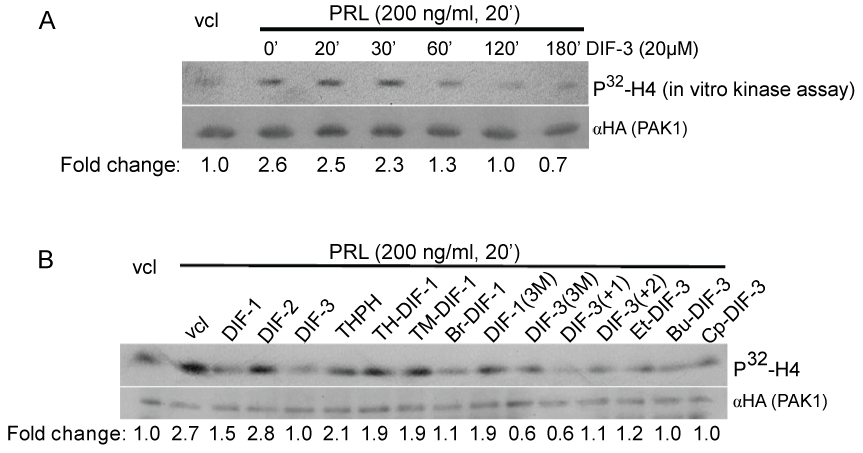
.
Figure 2: DIF-3 and its derivatives inhibit PAK1 kinase activity in cells.
A) MCF-7 cells over expressing HA-tagged PAK1 were serum-deprived, pre-treated with or without 20 μM DIF-3 over a time course and stimulated with or without PRL (200 ng/ml, 20 min) to activate PAK1. HA-PAK1 was immunoprecipitated with anti-HA Ab, subjected to an in vitro kinase assay using H4 histone as a substrate and probed with anti- HA Ab. Relative PAK1 kinase activity was then normalized by the amount of PAK1 immunoprecipitated in each lane. The numbers at the bottom give the relative fold
increase of 32P incorporation into H4 histone. The migrations of proteins are indicated.
B) MCF-7 PAK1 cells were serum-deprived, pre-treated for 2 h with vehicle or 20 μM DIFs and their derivatives as indicated, followed by PRL treatment (200 ng/ml, 20 min). in vitro kinase assay was performed as described in A.
View Figure 2
Next, we compared the inhibitory effect of some other DIF derivatives on PRL- dependent PAK1 kinase activity. The cells were incubated with the DIF derivatives (20 μM) for 2 h prior to PRL treatment. PAK1 kinase activity was inhibited by all the DIF derivatives to different degree with exception of DIF-2 and THPH (Figure 2B). THPH is a non-bioactive analog of DIF-1 which affects neither cell growth nor cell differentiation [5-8]. Both DIF-1 and DIF-3 exhibit anti-proliferative activities in different human cancer cells, as was described in the Introduction section, and indeed both DIF-1 and -3 inhibit PAK1 activity. Meanwhile, DIF-2, which does not inhibit proliferation in human leukemia K562 cells [5], also does not inhibit PAK1 in our assay. We have demonstrated that DIF-3 inhibits PAK1 stronger than DIF-1 (63% inhibition by DIF-3 vs. 44% by DIF-1) (Figure 2B). DIF-3 has been shown to exert a more powerful anti-proliferative effect than DIF-1 in human cervical cancer cells [10], leukemia [6,8] and colon cancer cells [12]. Interestingly, the order of inhibitory effect of DIFs on PAK1 in MCF-7 cells described herein corresponds to the order of their potency to inhibit proliferation of human leukemia K563 cells: DIF-3 > DIF-1 > DIF-2 and THPH [7]. Since DIF-2 was ineffective in PAK1 kinase assay, hexanone (the alkyl group in DIF-1) rather than pentanone in DIF-2 [7] appeared to be important for inhibition of PAK1. Because chemically modified derivatives of DIF-3 (Bu-DIF-3) has been shown to be a more potent anti-proliferative agent than DIF-3 [5], we tested the effect of different derivatives of DIF-1 and DIF-3 on PAK1 activity and demonstrated that DIF-3 derivatives DIF-3(3M) and DIF-3(+1) inhibit PAK1 kinase activity to the maximal degree in MCF-7 cells (78% inhibition) (Figure 2B).
Inhibition of PAK1 kinase activity by DIF-3(+1) is direct and ligand-independent
PAK1 is activated in response to PRL, estrogen and heregulin (a ligand for HER3 (human epidermal growth factor receptor-3) and HER4 (human epidermal growth factor receptor-4)) by different signaling pathways [32-34]. We decided to study whether the effect of DIF-3(+1), which has the strongest inhibitory effect of PAK1 activity, depends on a ligand. As demonstrated in Fig. 3A, PAK1 kinase activity was inhibited by DIF-3(+1) to the basal level in response to all three PRL, E2 and HRG (Figure 3A). These data suggests that DIF-3(+1) may act directly on PAK1 protein regardless of upstream effector.
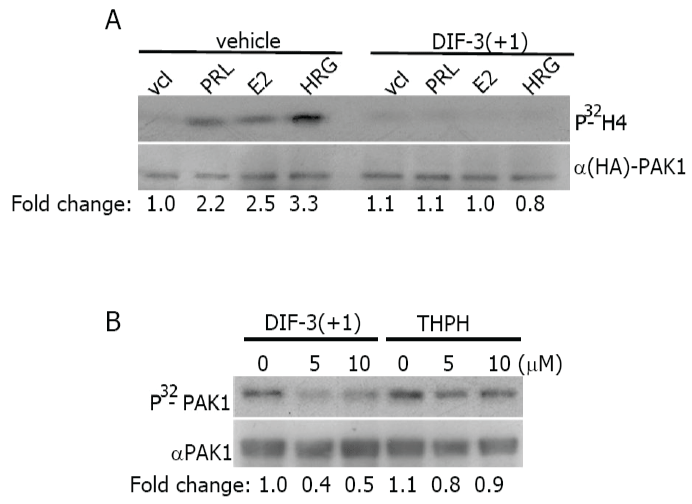
.
Figure 3: DIF-3(+1) inactivates purified PAK1 in vitro.
A) MCF-7 PAK1 cells were serum-deprived, pre-treated with vehicle or 20 μM DIF-3 (+1) for 2 h, and stimulated with or without PRL (200 ng/ml, 20 min), E2 (1 nM, 30 min) or HRG (30 ng/ml, 15 min). Immunoprecipitated HA-PAK1 was subjected to the in vitro kinase assay as described in Figure 2. Data are presented as in Figure 2B). Purified GST- tagged PAK1 was subjected to in vitro kinase assay in the presence of DIF-3(+1) or THPH. The same membrane was probed with anti-PAK1 antibody for detection of GST-
PAK1. Relative PAK1 kinase activity was then normalized by the amount PAK1 for each lane. The numbers at the bottom give the relative fold increase of 32P incorporation into PAK1.
View Figure 3
To determine whether DIF-3(+1) directly inhibits PAK1, exogenous purified GST-PAK1 was combined with DIF-3(+1) or with biologically inactive THPH and the level of PAK1 autophosphorylation was assessed by the in vitro kinase assay as described above but in the absence of H4 substrate. Both concentrations of DIF-3(+1), 5 μM and 10 μM, inhibited PAK1 autophosphorylation to the same degree while THPH had no effect (Figure 3B). These data suggest that DIF-3(+1) directly inhibits PAK1 in vitro.
DIF-3(+1) inhibits cyclin D1 promoter activity
Many factors, including PRL, E2, EGF, HRG, as well as activated PAK1 induce cyclin D1 expression ([35,36]; reviewed in [37]) while DIF-1 and DIF-3 decrease cyclin D1 expression level by inducing cyclin D1 proteolysis via activation of GSK-3β [10,12,13]. We have previously showed that, in response to PRL, tyrosyl phosphorylated PAK1 activates cyclin D1 promoter activity [30]. Therefore, we sought to determine whether DIF-3(+1)-dependent inhibition of PAK1 kinase activity plays a role in cyclin D1 regulation in response to PRL, E2, EGF and HRG. First, we measured the induction of cyclin D1 promoter activity in MCF-7 PAK1 cells treated with or without PRL, E2, EGF and HRG. As shown in Figure 4, the cells transfected with cyclin D1 promoter-luciferase construct demonstrated increased luciferase expression in response to PRL, E2, EGF and HRG as expected, with the greatest effect of HRG. However, when cells were treated with DIF-3(+1), induction of cyclin D1 promoter activity was significantly decreased for all four ligands studied (Figure 4). These data suggest that DIF-3(+1) negatively regulates the activity of the cyclin D1 promoter. PAK1 has been previously implicated in induction of cyclin D1 expression [21,36]. Balasenthil et al. proposed that active PAK1 can increase cyclin D1 transcription through two independent pathways-the NFκB pathway and phosphorylation of S305 of estrogen receptor alpha [21,36]. We have previously demonstrated that, in response to PRL, tyrosyl phosphorylated PAK1 translocates into the nucleus to activate cyclin D1 promoter activity and that adapter protein Nck keeps PAK1 into the cytoplasm, preventing activation of cyclin D1 promoter activity [30]. As DIF-3 induces nuclear translocation of GSK-3β [10], we may speculate that DIF-3(+1) affects intracellular localization of PAK1 to retain PAK1 in the cytoplasm leading to inhibition of cyclin D1 expression.
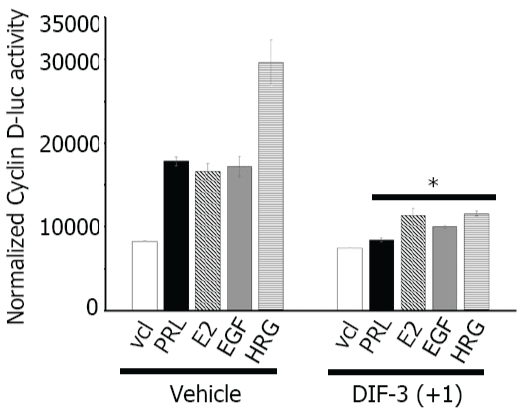
.
Figure 4: DIF-3(+1) decreases cyclin D1 promoter activity.
MCF-7 PAK1 cells were transfected with cyclin D1-luciferase reporter, serum-deprived and treated with vehicle or 5 μM DIF-3(+1) for 24 h in the presence of either vehicle, 200 ng/ml PRL, 1 nM E2, 10 ng/ml EGF or 30 ng/ml HRG. Luciferase activity was normalized with β-galactosidase activity. Bars represent mean ± S.E., *, p < 0.05 compare with cells in the same condition treated with vehicle, n = 3.
View Figure 4
DIF-3(+1) inhibits breast cancer cell proliferation
DIFs exhibit powerful anti- proliferative effect in various cancer cells [5-12]. However, the effect of DIFs on breast cancer cells has not been studied. In this study, we examined whether DIF-3(+1) inhibits the proliferation of breast cancer cells. First, to confirm the role of PAK1 in cell proliferation, we treated MCF-7 PAK1 and T47D PAK1 cells with cell- permeable small-molecule PAK1 inhibitor IPA-3 [38,39]. IPA-3 inhibited PRL- dependent proliferation of both MCF-7 and T47D cells (Figure 5A). Next, as shown in Figure 5B and C, PRL, E2, EGF and HRG induced proliferation of both MCF-7 and T47D cells to different degrees which probably depends on the amount of receptors to be activated by a ligand. We have shown that DIF-3(+1) strongly inhibited ligand-induced proliferation of both MCF-7 and T47D cells (Figure 5B and Figure 5C). Both cell lines were assessed for cytotoxicity for DIF-3(+1) which was not toxic (data not shown).
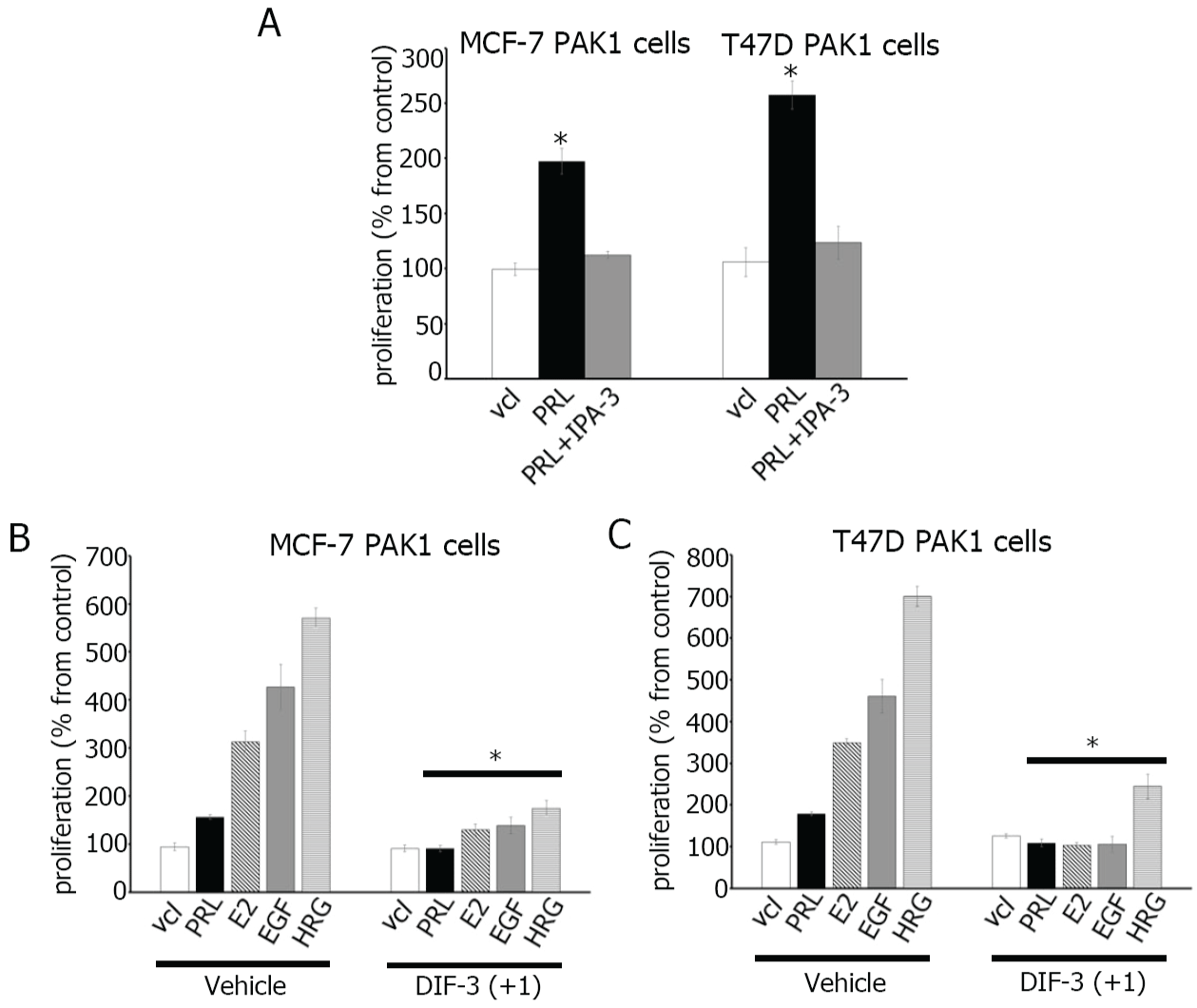
.
Figure 5: DIF-3(+1) inhibits breast cancer cell proliferation.
(A) MCF-7 and T47D cells overexpressing PAK1 were serum-deprived and incubated with vehicle (vcl), PRL (500 ng/ml) in presence or absence of IPA-3 (5 μM). MCF-7 (B) or T47D (C) cells overexpressing PAK1 were serum-deprived and incubated with vehicle or 5 μM DIF-3(+1) in the presence of either vehicle, 500 ng/ml PRL, 1 nM E2, 10 ng/ml EGF or 30 ng/ml HRG. Cell density was assessed after seven days by MTT cell proliferation assay. Changes in cell numbers at day 7 are shown as percentages of the vehicle-treated cell number. Bars represent mean ± S.E., *, p < 0.05 compare with cells in the same condition treated with vehicle, n = 3.
View Figure 5
Conclusion
Based on our data presented here and previous studies, we suggest that DIF-1 and -3 inhibit PAK1 kinase in cells. We have found that DIF-3(+1) derivative inhibits PAK1 kinase activity both in cells and in vitro that leads to negative regulation of cyclin D1 promoter activity and inhibition of cell proliferation. We propose PAK1 as a DIF-3(+1) target which mediates the anti-proliferative effect of DIF-3(+1) on breast cancer cells.
Acknowledgement
We thank Dr. Maruta (NF/TSC Cure Org., Melbourne, Australia) for his ideas and helpful discussion. This work was supported by the U.S. National Institutes of Health (Grant R01-DK88127 to MD).
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
References
-
Town CD, Gross JD, Kay RR (1976) Cell differentiation without morphogenesis in Dictyostelium discoideum. Nature 262: 717-719.
-
Morris HR, Taylor GW, Masento MS, Jermyn KA, Kay RR (1987) Chemical structure of the morphogen differentiation inducing factor from Dictyostelium discoideum. Nature 328: 811-814.
-
Kay RR, Berks M, Traynor D (1989) Morphogen hunting in Dictyostelium. Development 107 Suppl: 81-90.
-
Kay RR, Flatman P, Thompson CR (1999) DIF signalling and cell fate. Semin Cell Dev Biol 10: 577-585.
-
Gokan N, Kikuchi H, Nakamura K, Oshima Y, Hosaka K, et al. (2005) Structural requirements of Dictyostelium differentiation-inducing factors for their stalk-cell- inducing activity in Dictyostelium cells and anti-proliferative activity in K562 human leukemic cells. Biochem Pharmacol 70: 676-685.
-
Kubohara Y (1997) DIF-1, putative morphogen of D. discoideum, suppresses cell growth and promotes retinoic acid-induced cell differentiation in HL-60. Biochem Biophys Res Commun 236: 418-422.
-
Y. Kubohara (1999) Effects of differentiation-inducing factors of Dictyostelium discoideum on human leukemia K562 cells: DIF-3 is the most potent anti-leukemic agent. Eur J Pharmacol 381: 57-62.
-
Akaishi E, Narita T, Kawai S, Miwa Y, Sasaguri T, et al. (2004) Differentiation-inducing factor-1-induced growth arrest of K562 leukemia cells involves the reduction of ERK1/2 activity. Eur J Pharmacol 485: 21-29.
-
Shimizu K, Murata T, Tagawa T, Takahashi K, Ishikawa R, et al. (2004) Calmodulin-dependent cyclic nucleotide phosphodiesterase (PDE1) is a pharmacological target of differentiation-inducing factor-1, an antitumor agent isolated from Dictyostelium. Cancer Res 64: 2568-2571.
-
Takahashi-Yanaga F, Taba Y, Miwa Y, Kubohara Y, Watanabe Y, et al. (2003) Dictyostelium differentiation-inducing factor-3 activates glycogen synthase kinase-3beta and degrades cyclin D1 in mammalian cells. J Biol Chem 278: 9663-9670.
-
Kanai M, Konda Y, Nakajima T, Izumi Y, Kanda N, et al. (2003) Differentiation-inducing factor-1 (DIF-1) inhibits STAT3 activity involved in gastric cancer cell proliferation via MEK-ERK-dependent pathway. Oncogene 22: 548-554.
-
Kubokura N, Takahashi-Yanaga F, Arioka M, Yoshihara T, Igawa K, et al. (2015) Differentiation-inducing factor-3 inhibits intestinal tumor growth in vitro and in vivo. J Pharmacol Sci 127: 446-455.
-
Takahashi-Yanaga F, Yoshihara T, Jingushi K, Igawa K, Tomooka K, et al. (2014) DIF-1 inhibits tumor growth in vivo reducing phosphorylation of GSK-3β and expressions of cyclin D1 and TCF7L2 in cancer model mice. Biochem Pharmacol 89: 340-348.
-
Molli PR, Li DQ, Murray BW, Rayala SK, Kumar R (2009) PAK signaling in oncogenesis. Oncogene 28: 2545-2555.
-
Kumar R, Gururaj AE, Barnes CJ (2006) p21-activated kinases in cancer. Nat Rev Cancer 6: 459-471.
-
Eswaran J, Li DQ, Shah A, Kumar R (2012) Molecular pathways: targeting p21- activated kinase 1 signaling in cancer--opportunities, challenges, and limitations, Clin Cancer Res 18: 3743-3749.
-
Bekri S, Adélaïde J, Merscher S, Grosgeorge J, Caroli-Bosc F, et al. (1997) Detailed map of a region commonly amplified at 11q13-->q14 in human breast carcinoma. Cytogenet Cell Genet 79: 125-131.
-
Ong CC, Jubb AM, Haverty PM, Zhou W, Tran V, et al. (2011) Targeting p21-activated kinase 1 (PAK1) to induce apoptosis of tumor cells. Proc Natl Acad Sci USA 108: 7177-7182.
-
Holm C, Rayala S, Jirström K, Stål O, Kumar R, et al. (2006) Association between Pak1 expression and subcellular localization and tamoxifen resistance in breast cancer patients. J Natl Cancer Inst 98: 671-680.
-
Vadlamudi RK, Adam L, Wang RA, Mandal M, Nguyen D, et al. (2000) Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J Biol Chem 275: 36238-36244.
-
Balasenthil S, Sahin AA, Barnes CJ, Wang RA, Pestell RG, et al. (2004) p21-activated kinase-1 signaling mediates cyclin D1 expression in mammary epithelial and cancer cells. J Biol Chem 279: 1422-1428.
-
Salh B, Marotta A, Wagey R, Sayed M, Pelech S (2002) Dysregulation of phosphatidylinositol 3-kinase and downstream effectors in human breast cancer. Int J Cancer 98: 148-154.
-
Wang RA, Zhang H, Balasenthil S, Medina D, Kumar R (2006) PAK1 hyperactivation is sufficient for mammary gland tumor formation. Oncogene 25: 2931-2936.
-
Li Q, Mullins SR, Sloane BF, Mattingly RR (2008) p21-Activated kinase 1 coordinates aberrant cell survival and pericellular proteolysis in a three-dimensional culture model for premalignant progression of human breast cancer. Neoplasia 10: 314-329.
-
Arias-Romero LE, Villamar-Cruz O, Pacheco A, Kosoff R, Huang M, et al. (2010) A Rac-Pak signaling pathway is essential for ErbB2- mediated transformation of human breast epithelial cancer cells. Oncogene 29: 5839-5849.
-
Arias-Romero LE, Villamar-Cruz O, Huang M, Hoeflich KP, Chernoff J (2013) Pak1 kinase links ErbB2 to ß-catenin in transformation of breast epithelial cells. Cancer Res 73: 3671-3682.
-
Hammer A, Diakonova M (2015) Tyrosyl phosphorylated serine-threonine kinase PAK1 is a novel regulator of prolactin-dependent breast cancer cell motility and invasion. Adv Exp Med Biol 846: 97-137.
-
Hammer A, Oladimeji P, De Las Casas LE, Diakonova M (2015) Phosphorylation of tyrosine 285 of PAK1 facilitates betaPIX/GIT1 binding and adhesion turnover. FASEB J 29: 943-959.
-
Hammer A, Rider L, Oladimeji P, Cook L, Li Q, et al. (2013) Tyrosyl phosphorylated PAK1 regulates breast cancer cell motility in response to prolactin through filamin A. Mol Endocrinol 27: 455-465.
-
Tao J, Oladimeji P, Rider L, Diakonova M (2011) PAK1-Nck regulates cyclin D1 promoter activity in response to prolactin. Mol Endocrinol 25: 1565-1578.
-
Rider L, Oladimeji P, Diakonova M (2013) PAK1 regulates breast cancer cell invasion through secretion of matrix metalloproteinases in response to prolactin and three-dimensional collagen IV. Mol Endocrinol 27: 1048-1064.
-
Rider L, Shatrova A, Feener EP, Webb L, Diakonova M (2007) JAK2 tyrosine kinase phosphorylates PAK1 and regulates PAK1 activity and functions. J Biol Chem 282: 30985-30996.
-
Mazumdar A, Kumar R (2003) Estrogen regulation of Pak1 and FKHR pathways in breast cancer cells. FEBS Lett 535: 6-10.
-
Adam L, Vadlamudi R, Kondapaka SB, Chernoff J, Mendelsohn J, et al. (1998) Heregulin regulates cytoskeletal reorganization and cell migration through the p21-activated kinase-1 via phosphatidylinositol-3 kinase. J Biol Chem 273: 28238-28246.
-
Brockman JL, Schroeder MD, Schuler LA (2002) PRL activates the cyclin D1 promoter via the Jak2/Stat pathway. Mol Endocrinol 16: 774-784.
-
Balasenthil S, Barnes CJ, Rayala SK, Kumar R (2004) Estrogen receptor activation at serine 305 is sufficient to upregulate cyclin D1 in breast cancer cells. FEBS Lett 567: 243-247.
-
Fu M, Wang C, Li Z, Sakamaki T, Pestell RG (2004) Minireview: Cyclin D1: normal and abnormal functions. Endocrinology 145: 5439-5447.
-
Deacon SW, Beeser A, Fukui JA, Rennefahrt UE, Myers C, et al. (2008) An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem Biol 15: 322-331.
-
J Viaud, JR Peterson (2009) An allosteric kinase inhibitor binds the p21-activated kinase autoregulatory domain covalently. Mol Cancer Ther 8: 2559-2565.
