

International Journal of Cancer and Clinical Research
ADAM17: A Gatekeeper in Immune-Oncology?
Peter R Lowe* and Nathalie Corvaia
Centre d'Immunologie Pierre Fabre, Laboratoire Pierre Fabre, France
*Corresponding author:
Peter R Lowe, Department of mAbOptimization, Centre d'Immunologie Pierre Fabre, 5 Avenue Napoleon III, 74160, St Julien en Genevois, France, E-mail: peter.lowe@pierre-fabre.com
Int J Cancer Clin Res, IJCCR-3-058, (Volume 3, Issue 3), Mini Review; ISSN: 2378-3419
Received: April 13, 2016 | Accepted: June 17, 2016 | Published: June 20, 2016
Citation: Lowe PR, Corvaia N (2016) ADAM17: A Gatekeeper in Immune-Oncology? Int J Cancer Clin Res 3:058. 10.23937/2378-3419/3/3/1058
Copyright: © 2016 Lowe PR, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Recent therapeutics searching to reactivate and target the immune system to destroy tumors have demonstrated remarkable success in the treatment of patients with previously intractable disease such as metastatic melanoma. Current research is enlarging the spectrum of targets and strategies for enhancing the immune response against tumors, in order to further improve treatment efficacy. In that respect, ADAM17, has frequently been described for it's over expression or over activation in the tumor microenvironment. ADAM17 is responsible for the shedding of a large number of receptors and ligands involved in immune tumor targeting and the deregulation of this activity should be considered when developing therapeutic strategies. The aim of this review is to summarize the spectrum of ADAM17 substrates and its potential role in tumor immune modulation.
Keywords
ADAM17, Shedding, Sheddase, Immunotherapy
Introduction
A disintegrin and metalloprotease 17 (ADAM17) a cell surface protease of the metzincin family was discovered in 1997 as the protein responsible for the extracellular cleavage or shedding of TNF-α and was originally named TNF-α converting enzyme (TACE) [1]. A further twenty ADAMs have since been described in humans albeit that only thirteen have functional protease domains [2]. ADAM17 and the closely related ADAM10 have been the most widely studied due to the large range and often overlapping substrate specificities of the two enzymes [2] and in particular due to their association with pathologies including cancer, inflammatory disorders and Alzheimer's disease [3]. The biological activity of the two enzymes differ however, ADAM10 being constitutively active [4,5], whereas ADAM17 is an inducible sheddase [5-8]. The inducible nature of ADAM17 shedding activity corresponds to its role in inflammatory responses [9-12] and wound healing [13,14] responding to various upstream signals including lysophasphatidic acid (LPA) [15], bradykinin [16], lipopolysaccharide (LPS) [17] and through the activation of different protein kinase C sub classes [18,19].
The role of ADAM17 in the release of soluble TNF-α led to considerable interest and numerous clinical trials have been conducted of small molecule inhibitors for the treatment of rheumatoid arthritis [20], however, lack of specificity and toxicity coupled with the development of TNF targeting immunotherapies has slowed further clinical development. The dual specificity small molecule inhibitor of ADAM17/ADAM10, INCB7839, was evaluated in phase II clinical trials for the treatment of Trastuzumab resistant breast cancer [21], and demonstrated reduced shedding of a number of evaluated substrates [22].
ADAM17 is the major inducible sheddase for the ErbB ligands; Amphiregulin [23], TGF-α [24], Epiregulin [25], Epigen [26], HB-EGF [27] and certain members of the Neuregulin family [28], and thus implicates itself in a wide range of developmental and repair pathways. It is indeed the excessive shedding of certain ligands, in particular Amphiregulin [29], HB-EGF [30] and TGF-α [31] that are associated with a wide range of cancer pathologies through the downstream effect upon the ErbB receptors and in particular EGFR. The therapeutic potential for targeting ADAM17 in this context has been considered previously in excellent review articles and thus will not be repeated here [3,32,33]. Beyond the shedding of growth factors and the direct impact on neoplastic growth, ADAM17 is involved in the shedding of a broad range of factors implicated in the activation, recruitment and resolution of innate and adaptive immune responses. The shedding of these different factors is an intrinsic mechanism in the control of the immune and inflammatory response, however, in certain cancers this may be deregulated through the over expression [34-37] or over activation [38] of ADAM17.
The implication of the immune system in tumor surveillance and destruction has been a subject of debate for decades since the original hypothesis of immunosurveillance was proposed by Burnet [39,40] and Thomas [41]. More recently the concepts of T-cell exhaustion [42] and tumor immune evasion [43] have been added to this complex game of cat and mouse. The study of immune modulation has recently yielded new therapeutic options for health care practitioners and patients, through the approval of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) targeted by Ipilimumab [44] and more recently programmed cell death 1 (PD-1) targeted by Nivolumab [45] and Pembrolizumab [46]. These treatments have been approved for the use in the treatment of metastatic melanoma, Nivolumab also being approved for use in squamous non-small cell lung cancer (NSCLC) [47,48]. These targeted therapies represent only the first in a raft of products currently under evaluation that offer great potential to reactivate a suppressed or exhausted immune system. The number of receptors and ligands that mediate the migration, activation and maintenance of inflammatory and immune responses essential to the immune surveillance machinery that have been identified as substrates for ADAM17 is already long and probably incomplete.
The following sections will expand on the implications of the deregulated shedding by ADAM17 of immune regulatory substrates in the tumor environment.
NK Cell Tumor Targeting
Epithelial tumors have been found to express MHC class I chain–related molecules (MICs) [49] that act as ligands for NKG2D on γδ and CD8+αβ T cells and NK cells and elicit cytolytic targeting of the tumors [50]. The presentation of MICA and MICB results from the stress induced state of the tumor and targets these cells for destruction. Elevated levels of MICA/B have been described in a wide range of solid tumors including breast, lung, ovarian, prostate, renal and colon carcinomas [51]. Further investigation demonstrated that tumors positive for MICA/B expression demonstrated a 60-70% decrease of the NKG2D expression in tumor infiltrating lymphocytes (TILs) and peripheral blood mononuclear cells (PBMCs) and this is believed to contribute to the loss of NKG2D mediated tumor targeting [52]. Tumors positive for MIC expression demonstrated measurable levels of soluble MIC in the serum of patients that is absent in the serum of MIC-tumors. The protein disulfide isomerase ERp5 [53,54] has been shown to modulate the configuration of cell surface MICA/B and renders them susceptible to shedding by ADAM17 and to a lesser extent ADAM10 [50,55]. The extracellular shedding of MICA and MICB is not exclusive to ADAM17 and ADAM10 and indeed their secretion in exosomes has been observed in certain tumor cell lines [55] thus the specific targeting of MICA and or B may provide a more precise therapeutic outcome [56] than inhibiting their shedding. The NKG2D ligands expressed on tumors are not restricted to the MIC as indeed UL16 binding proteins (ULBP) are also presented and shed from tumors. The presentation and shedding of NKG2D ligands be they MIC or ULBPs represents a primary mechanism of lymphocyte tumor targeting, however, shedding is not the only regulatory device controlling this interaction, down regulation of the receptor or the ligand by a myriad of strategies can have the same net effect, these alternative strategies were detailed in a recent review [57].
Recently an additional tumor expressed ligand for NK cell engagement has been described, a member of the B7 family of immune regulatory proteins, B7-H6 [58]. Tumor expressed B7-H6 has been shown to engage with the activating receptor NKp30 on NK cells to enhance anti tumoral cellular. Preclinical evidence suggests that T-cell recruitment through B7-H6 addressed BiTes [59] and NK recruitment via bispecific tumor antigen/B7-H6 fusion proteins [60] demonstrate that the potential of B7-H6 to recruit an anti-tumor immune response is significant. The presentation of B7-H6 at the tumor cell surface is controlled as with MICA/B through extracellular shedding by ADAM17 and ADAM10, siRNA inhibition implicating a greater role for ADAM17 [61]. The detection of elevated levels of soluble B7-H6 in the serum of a subset of melanoma patients [61] suggests that NK cell targeting via B7-H6 can be silenced through the down regulation of NKp30 similar to the effect observed with MICA/B and NKG2D and may further contribute to the attenuation of innate anti tumoral cellular cytotoxicity. The full scope of proteases capable of shedding B7-H6 has yet to be fully explored, however, at present the targeted inhibition of ADAM17 represents a potentially important mechanism to reactivate B7-H6 mediated NK cell tumor targeting.
Fas/FasL and ADAM17
The Fas receptor (Fas, CD95) /Fas ligand (FasL, CD95L) interaction is a key mediator of cellular apoptosis and is essential to the regulation of the adaptive immune response, failures in this system resulting in autoimmune lymphoproliferative syndrome [62]. In the tumor environment compromised Fas signaling renders cells resistant to immune surveillance and induced apoptosis [63], the expression of Fas and FasL by tumor cells being shown to promote tumor growth [64]. The tumor counter attack hypothesis promotes the possibility that the expression of FasL by tumors results in the apoptosis of infiltrating lymphocytes either through cell contact or the release of FasL presenting microvesicles [65]. At present the distribution of FasL on certain tumor types appears ambiguous [66] and its activity when present has been described as either the induction of T-cell apoptosis or the down regulation of the T-cell receptor(TCR) component CD3ε [67]. ADAM10 has been shown to mediate the constitutive shedding of FasL from T lymphocytes [5] potentially maintaining lymphocyte homeostasis, whereas ADAM17 has been shown to be rapidly up regulated upon cellular activation along with recruitment of FasL to the cell surface [68]. The deregulated shedding activity of ADAM17 described in the tumor environment coupled with tumor expressed FasL create the potential for a potent inhibition of T-cell induced apoptosis of tumor cells through competition for Fas binding.
Antibody Dependent Cellular Cytotoxicity Regulation
Perhaps of immediate clinical relevance is the capacity of immunotherapies targeting tumors to illicit antibody dependent cellular cytotoxicity (ADCC) such as is observed with Her2 targeting Trastuzumab and Pertuzumab [69] and the CD20 targeting Rituximab [70]. Antibody bound to its target tumor antigen ligates the FcγRIII and invokes cytokine release and degranulation of NK cells. Two isoforms of FcγRIII exist CD16a and CD16b, the first a trans membrane protein the second attached to the membrane via a glycophosphatidylinositol anchor. CD16a is found predominantly on NK cells [71], whereas CD16b is found exclusively on neutrophils [72]. Extensive research has been performed to enhance antibody CD16a activation of cellular cytotoxicity through modulation of the primary protein sequence of the antibody or modification of the post translational glycosylation [73]. Obinutuzumab a monoclonal antibody targeting CD20 developed by GlycArt/Roche that relies on reduced fucosylation to enhance its ADCC activity was the first approved antibody using enhanced ADCC for the treatment of chronic lymphocytic leukemia [74]. The ligation of CD16a to tumor bound antibody Fc domains results in the release of INFγ and TNFα from NK cells, however it has recently been observed that this is accompanied by a proportional loss of CD16a and the adhesion molecule L-selectin (CD62L) [71]. Inhibiting ADAM17 was shown to reduce CD16a and CD62L shedding from NK cells upon cytokine activation or through Rituximab ligation to Raji cells [75]. The inhibition of ADAM17 also provoked an increase in intracellular INFα and TNFα in the activated NK cells although there was no increase in Raji cell lysis [76].
The potential therapeutic impact of inhibiting ADAM17 in this context remains unclear as no enhanced cell lysis by ADCC was observed, however, the increased levels of TNFα and INFγ may indeed impact the immune surveillance response in a heterogeneous and intact lymphatic system. The murine CD16 orthologue is not shed [75] and thus limits the study of this potential activation at present. The significance of CD62L shedding will be considered in a later section of this review.
Neutrophil Mediated Antibody Targeted Tumor Destruction
The efficacy of therapeutic monoclonal antibodies in the elimination of tumors is largely attributed to their direct effect on the target antigen and the induced elimination by ADCC and Complement Dependent Cytotoxicity (CDC), somewhat less well defined is the implication of Neutrophils in the elimination of tumors. Indeed the observation that elevated neutrophil to lymphocyte ratios in cancer patients is prognostic for shorter overall survival [77] an effect suspected to be mediated via the maintenance of a chronic inflammatory state and the production of angiogenic factors. None the less it has recently been demonstrated that neutrophils alone, at least in the experimental conditions tested, were sufficient to mediate an antibody targeted anti tumoral immune response [78]. The Fcγ receptor CD16b is shed from the cell surface of neutrophils by ADAM17 following cell activation via IgG immune complex or during apoptosis. The cleavage site of CD16a and b have been shown to be conserved and are located in the membrane proximal domain of each protein [75] despite the different membrane attachment mechanisms of these receptors. Circulating levels of soluble CD16b are high due presumably to the high daily turnover of neutrophils by apoptosis, in the order of 10 [11] cells per day in a healthy adult [72]. The targeted inhibition of ADAM17 and ADAM10 in patients treated with INCB7839 demonstrated an almost two fold decrease in circulating CD16 [72] although the study did not discriminate between CD16a and b. Selective inhibition of ADAM17 and ADAM10 was able to determine that ADAM17 is principally responsible for the shedding of CD16b from neutrophils. As with NK cells, neutrophils shed CD62L via ADAM17 [79], and conditional knock out models demonstrated that the loss of ADAM17 resulted in up to ten times more CD62L on neutrophils, resulting in slow rolling [79] and more rapid infiltration to sites of inflammation [9,79], albeit that overall infiltrating neutrophil levels were lower [9]. Soluble CD62L has been described at relatively high levels in normal serum, the mean value determined as 1.6 μg/ml [80], in vitro experiments demonstrated that this physiological level was already sufficient to reduce leukocyte binding.
Adaptive and Innate Immune Targeting of Tumors
Whereas NK cells have a range of targeting receptors that recognize damaged or infected cells that are subsequently targeted for destruction, the adaptive cellular immune response functions through a separate system of cellular markers and receptors that normally differentiate self from non-self and in the case of tumor immune targeting recognize aberrant cell surface markers including phosphatidylserine or major his to compatibility (MHC) presented tumor specific peptides. The functioning of the T cell response to tumors has been recently reviewed [81] as has the exhaustion of T cells that leads to tumor progression [82] and how current immune checkpoint inhibitors are redressing this [83], and as such will not be discussed here. There are however, a number of additional immune checkpoints that are involved in T cell tumor targeting that are once again targets for shedding.
Activated T cells and NK cells express Lymphocyte-activation gene 3 (LAG-3, CD223), a receptor that binds MHC class II proteins on antigen presenting cells (APCs) [84] and is a negative regulator of activated T cell proliferation and cytokine production. LAG3 has been shown to be constitutively shed from the surface of cells by ADAM10 [84] the absence of constitutive LAG-3 shedding through ADAM10 knock down or the expression of non-cleavable LAG-3 resulting in reduced T cell proliferation or cytokine release [84]. ADAM17 shedding of LAG-3 has been shown to be induced as a result of T cell receptor (TCR) activation via protein kinase θ signalling. T cell activation has been shown to increase 12 fold the level of shedding of LAG-3 [84]. Myeloid derived suppressor cells (MDSC) have recently been shown to up regulate cellular LAG-3 levels in exhausted CD4+ and CD8+ T cells [85], although it was not described in this study if the shedding of LAG-3 was modulated.
The inhibitory immune check point receptor, T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) is expressed on effector and regulatory T cells, tumor associated dendritic cells (TADCs) and macrophages [86,87]. The ligand for TIM-3, the C type lectin galectin-9, itself expressed on tumor cells and MDSCs, induces cell death and exhaustion of CD8+ T cells. TADC expressed TIM-3 has also been shown to bind free high mobility group protein 1 (HMGB1) preventing the transport and presentation of DNA released from dying tumor cells and preventing activation of the innate immune response [87]. TIM-3 has recently been identified as a substrate for ADAM17 and ADAM10 [86] with ADAM10 being demonstrated as the physiologically induced sheddase of TIM-3 in LPS activated CD14+ monocytes. The precise role of TIM-3 shedding in the attenuation or activation of an anti tumoral immune response remains to be further qualified as indeed would the impact of inhibiting over active ADAM17 in the tumor microenvironment.
In addition to TIM-3 the remaining human TIMs 1 and 4 have also been described as substrates for ADAM17 [88]. Expression of TIM-1 has been described on a number of immune cell types including activated T cells (preferentially Th2 cells), mast cells, natural killer cells, dendritic cells and B cells, the expression of TIM-4 is restricted to antigen presenting cells [88]. TIM-1 and 4 serve as phosphatidylserine receptors to engulf apoptotic cells, they are also known to interact with each other and regulate T cell proliferation [89]. The shed forms of TIM-1 and 4 are still capable of binding phosphatidylserine and it has been suggested that the presence of soluble receptors may inhibit engulfment and potentially attenuate an anti tumoral response through the non-proliferation of T helper cells and presentation of tumor antigens by dendritic cells.
In addition to TIM-3 macrophages express CD36 a receptor for phosphatidylserine that is necessary for efficient efferocytosis and the resolution of inflammation [90]. ADAM17 has been demonstrated to shed CD36 from macrophages, inhibition of this shedding leading to enhanced efferocytosis. The inflammatory state of the tumor microenvironment is considered a hallmark of tumor development [91] and the inflammatory state is dependent at least in part on ADAM17 mediated shedding of TNFα from macrophages [92]. The elevated shedding of TNFα in parallel to CD36 due to aberrant ADAM17 activation would promote the inflammatory state and prevent the removal of phosphatidylserine presenting cells, the inhibition of this shedding would potential resolve both of these situations.
A final activating receptor ligand pair in the form of CD40/CD40L is also involved in the activation of T cells that would ordinarily participate in the targeting of tumor cells for destruction by the immune system. Upon ligation of CD40L to its receptor CD40 pro inflammatory signals are induced with the concomitant shedding of CD40L by ADAM17 or ADAM10, the precise nature of the sheddase involved being cell type dependant [93]. The shedding of CD40 by ADAM17 has also been described in hemodialysis patients [94] and thus creates perhaps the only ligand receptor pairing for which ADAM17 regulates the shedding of both receptor and ligand. Soluble CD40L is capable of binding CD40, however, with greatly attenuated capacity to activate and thus this shedding event potentially attenuates normal T cell activation to prevent over activation. The heightened activity of ADAM17 in the tumor microenvironment may, through elevated CD40L shedding, prevent the successful activation of CD40. The development of CD40 agonists may serve to compensate this deficit although consideration should be given to the shedding of CD40 itself and its potential impact upon this therapeutic approach (Figure 1).
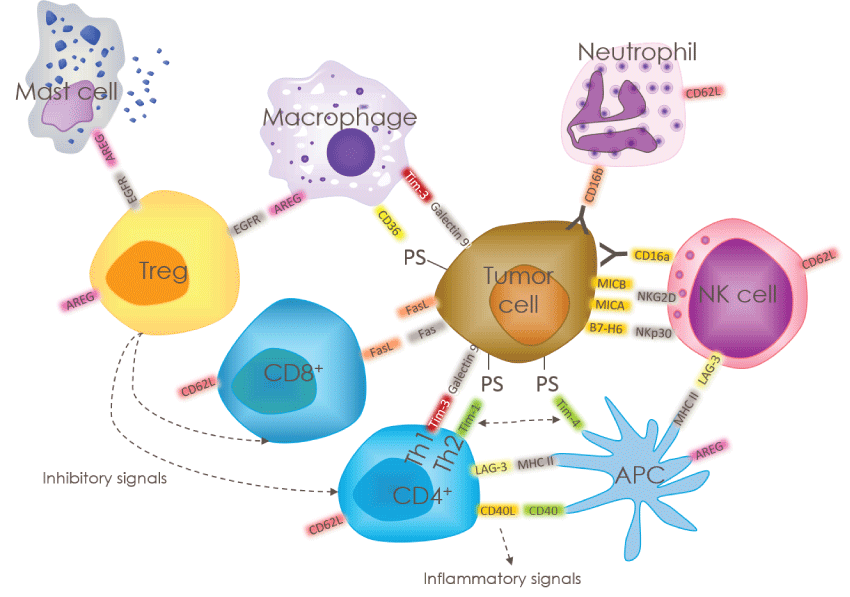
.
Figure 1: Substrates of ADAM17 in the tumor microenvironment. Substrates of ADAM17 are indicated in colored, molecules not shed by ADAM17 are in grey. CD62L; L-seclectin, CD16b; Fc gamma receptor III B, CD16a; Fc gamma receptor IIIA, MICA/B; MHC class I chain–related molecule A/B, NKG2D; Killer cell lectin-like receptor subfamily K member 1, B7-H6; Natural cytotoxicity triggering receptor 3 ligand 1, NKp30; Natural cytotoxicity triggering receptor 3, LAG-3; Lymphocyte activation gene 3 protein, MHCII; HLA class II histocompatibility antigen, AREG; Amphiregulin, Tim-1/3/4; T-cell immunoglobulin and mucin domain-containing protein 1/3/4, CD40; Tumor necrosis factor receptor superfamily member 5, CD40L; CD40 ligand, Fas; Tumor necrosis factor receptor superfamily member 6, FasL; Tumor necrosis factor ligand superfamily member 6, CD36; Platelet glycoprotein 4.
View Figure 1
T Regulatory Cells
Although no receptor or ligand pairs have currently been identified that influence the proliferation or activation of regulatory T (Treg) cells to inhibit the antitumoral immune response, a more classical proliferative mechanism that is itself dependent upon ADAM17 has been described. Tregs infiltrate and proliferate in to the tumor microenvironment and participate in the attenuation of the anti tumoral immune response. Tregs express the epidermal growth factor receptor (EGFR) and it has been shown that Amphiregulin (AREG) can induce proliferation and activation of the T-cell suppressive functions [95] of Tregs. AREGis produced by multiple tumor types [29,96], but also immune cells including mast cells [95], macrophages [97], dendritic cells [98] and Tregs themselves [99], ADAM17 being responsible for the shedding of the functional serum form. Targeting the EGFR receptor has demonstrated therapeutic efficacy, the possibilityof targeting the ligands of EGFR including AREG have also been explored as therapeutic targets [100-103]. The inhibition of AREG shedding may in particular offer the additional advantage of reducing Treg activity.
Conclusion
A wide range of immunotherapies are currently being developed to enhance the capacity of the immune system to through the activation of receptors such as CD40 or the inhibition of others such as TIM-3. A second family seeks to enhance the formation of immunological synapses between immune and tumor cells through the engagement of tumor antigens and immune receptors such as NKG2D. It is worthy of note that the natural regulation of a wide range of these systems for immune tumor targeting and activation are under the control of a small number of regulatory sheddases. The sheddase is itself, in particular ADAM17, a potential source of deregulation in the tumor environment and adds an additional level of complexity to the manipulation of these natural systems. When one considers the natural role of ADAM17 is restricted to the wound healing response and the resolution of inflammation it's therapeutic targeting when over activated in the tumor environment either alone or in combination with other immune modulating therapies merit investigation.
References
-
Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, et al. (1997) A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385: 729-733.
-
Edwards DR, Handsley MM, Pennington CJ (2008) The ADAM metalloproteinases. Mol Aspects Med 29: 258-289.
-
Arribas J, Esselens C (2009) ADAM17 as a therapeutic target in multiple diseases. Curr Pharm Des 15: 2319-2335.
-
Anderegg U, Eichenberg T, Parthaune T, Haiduk C, Saalbach A, et al. (2009) ADAM10 is the constitutive functional sheddase of CD44 in human melanoma cells. J Invest Dermatol 129: 1471-1482.
-
Ebsen H, Schroder A, Kabelitz D, Janssen O (2013) Differential surface expression of ADAM10 and ADAM17 on human T lymphocytes and tumor cells. PLoS One 8: e76853.
-
Doedens JR, Mahimkar RM, Black RA (2003) TACE/ADAM-17 enzymatic activity is increased in response to cellular stimulation. Biochem Biophys Res Commun 308: 331-338.
-
Gschwind A, Hart S, Fischer OM, Ullrich A (2003) TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J 22: 2411-2421.
-
Gooz M (2010) ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol 45: 146-169.
-
Arndt PG, Strahan B, Wang Y, Long C, Horiuchi K, et al. (2011) Leukocyte ADAM17 regulates acute pulmonary inflammation. PLoS One 6: e19938.
-
Brennan FM, Green P, Amjadi P, Robertshaw HJ, Alvarez-Iglesias M, et al. (2008) Interleukin-10 regulates TNF-alpha-converting enzyme (TACE/ADAM-17) involving a TIMP-3 dependent and independent mechanism. Eur J Immunol 38: 1106-1117.
-
Chalaris A, Garbers C, Rabe B, Rose-John S, Scheller J (2011) The soluble Interleukin 6 receptor: generation and role in inflammation and cancer. Eur J Cell Biol 90: 484-494.
-
Chalaris A, Adam N, Sina C, Rosenstiel P, Lehmann-Koch J, et al. (2010) Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J Exp Med 207: 1617-1624.
-
Yin J, Yu FS (2009) ERK1/2 mediate wounding- and G-protein-coupled receptor ligands-induced EGFR activation via regulating ADAM17 and HB-EGF shedding. Invest Ophthalmol Vis Sci 50: 132-139.
-
Kawaguchi M, Suzuki T (2014) Gene expression and in situ localization of ADAM17 during skin wound healing. Int J Dermatol 53: e229-231.
-
Braun AH, Coffey RJ (2005) Lysophosphatidic acid, a disintegrin and metalloprotease-17 and heparin-binding epidermal growth factor-like growth factor in ovarian cancer: the first word, not the last. Clin Cancer Res 11: 4639-4643.
-
Dey M, Baldys A, Sumter DB, Gooz P, Luttrell LM, et al. (2010) Bradykinin decreases podocyte permeability through ADAM17-dependent epidermal growth factor receptor activation and zonula occludens-1 rearrangement. J Pharmacol Exp Ther 334: 775-783.
-
Ali M, Saroha A, Pewzner-Jung Y, Futerman AH (2015) LPS-mediated septic shock is augmented in ceramide synthase 2 null mice due to elevated activity of TNFα-converting enzyme. FEBS Lett 589: 2213-2217.
-
Kveiborg M, Instrell R, Rowlands C, Howell M, Parker PJ (2011) PKCα and PKCδ regulate ADAM17-mediated ectodomain shedding of heparin binding-EGF through separate pathways. PLoS One 6: e17168.
-
Alfa Cissé M, Louis K, Braun U, Mari B, Leitges M, et al. (2008) Isoform-specific contribution of protein kinase C to prion processing. Mol Cell Neurosci 39: 400-410.
-
Das Gupta S, Murumkar PR, Giridhar R, Yadav MR (2009) Current perspective of TACE inhibitors: a review. Bioorg Med Chem 17: 444-459.
-
Katharina Feldinger, Daniele Generali, Gabriela Kramer-Marek, Merel Gijsen, Tzi Bun Ng, et al. (2009) Clinical Benefit of INCB7839, a Potent and Selective Inhibitor of ADAM10 and ADAM17, in Combination with Trastuzumab in Metastatic HER2 Positive Breast Cancer Patients. Cancer Res 69: 5056.
-
Feldinger K, Generali D, Kramer-Marek G, Gijsen M, Ng TB, et al. (2014) ADAM10 mediates trastuzumab resistance and is correlated with survival in HER2 positive breast cancer. Oncotarget 5: 6633-6646.
-
Sunnarborg SW, Hinkle CL, Stevenson M, Russell WE, Raska CS, et al. (2002) Tumor necrosis factor-alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J Biol Chem 277: 12838-12845.
-
Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, et al. (1998) An essential role for ectodomain shedding in mammalian development. Science 282: 1281-1284.
-
Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, et al. (2004) Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol 164: 769-779.
-
Sahin U, Blobel CP (2007) Ectodomain shedding of the EGF-receptor ligand epigen is mediated by ADAM17. FEBS Lett 581: 41-44.
-
Merlos-Suárez A, Ruiz-Paz S, Baselga J, Arribas J (2001) Metalloprotease-dependent protransforming growth factor-alpha ectodomain shedding in the absence of tumor necrosis factor-alpha-converting enzyme. J Biol Chem 276: 48510-48517.
-
Montero JC, Yuste L, Díaz-Rodríguez E, Esparís-Ogando A, Pandiella A (2000) Differential shedding of transmembrane neuregulin isoforms by the tumor necrosis factor-alpha-converting enzyme. Mol Cell Neurosci 16: 631-648.
-
Berasain C, Avila MA (2014) Amphiregulin. Semin Cell Dev Biol 28: 31-41.
-
Kasai N, Kobayashi K, Shioya S, Yoshikawa Y, Yotsumoto F, et al. (2012) Soluble heparin-binding EGF-like growth factor (HB-EGF) detected by newly developed immuno-PCR method is a clear-cut serological biomarker for ovarian cancer. Am J Transl Res 4: 415-421.
-
Baselga J, Mendelsohn J, Kim YM, Pandiella A (1996) Autocrine regulation of membrane transforming growth factor-alpha cleavage. J Biol Chem 271: 3279-3284.
-
Murphy G (2008) The ADAMs: signalling scissors in the tumour microenvironment. Nat Rev Cancer 8: 929-941.
-
Scheller J, Chalaris A, Garbers C, Rose-John S (2011) ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol 32: 380-387.
-
Blanchot-Jossic F, Jarry A, Masson D, Bach-Ngohou K, Paineau J, et al. (2005) Up-regulated expression of ADAM17 in human colon carcinoma: co-expression with EGFR in neoplastic and endothelial cells. J Pathol 207: 156-163.
-
Lendeckel U, Kohl J, Arndt M, Carl-McGrath S, Donat H, et al. (2005) Increased expression of ADAM family members in human breast cancer and breast cancer cell lines. J Cancer Res Clin Oncol 131: 41-48.
-
Ni SS, Zhang J, Zhao WL, Dong XC, Wang JL (2013) ADAM17 is overexpressed in non-small cell lung cancer and its expression correlates with poor patient survival. Tumour Biol 34: 1813-1818.
-
Wu K, Liao M, Liu B, Deng Z (2011) ADAM-17 over-expression in gallbladder carcinoma correlates with poor prognosis of patients. Med Oncol 28: 475-480.
-
Pruessmeyer J, Martin C, Hess FM, Schwarz N, Schmidt S, et al. (2010) A disintegrin and metalloproteinase 17 (ADAM17) mediates inflammation-induced shedding of syndecan-1 and -4 by lung epithelial cells. J Biol Chem 285: 555-564.
-
Burnet M (1957) Cancer; a biological approach. I. The processes of control. Br Med J 1: 779-786.
-
Burnet FM (1970) The concept of immunological surveillance. Prog Exp Tumor Res 13: 1-27.
-
Thomas L (1959) Cellular and Humoral Aspects of the Hypersensitive States. Hoeber-Harper, New York.
-
Yi JS, Cox MA, Zajac AJ (2010) T-cell exhaustion: characteristics, causes and conversion. Immunology 129: 474-481.
-
Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, et al. (2015) Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol 35 Suppl: S185-198.
-
Cameron F, Whiteside G, Perry C (2011) Ipilimumab: first global approval. Drugs 71: 1093-1104.
-
Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, et al. (2015) Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 16: 375-384.
-
Robert C, Schachter J, Long GV, Arance A, Grob JJ, et al. (2015) Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 372: 2521-2532.
-
Sundar R, Cho BC, Brahmer JR, Soo RA (2015) Nivolumab in NSCLC: latest evidence and clinical potential. Ther Adv Med Oncol 7: 85-96.
-
West H (2014) Nivolumab as first line monotherapy for advanced non-small cell lung cancer: could we replace first line chemotherapy with immunotherapy? Transl Lung Cancer Res 3: 400-402.
-
Bauer S, Groh V, Wu J, Steinle A, Phillips JH, et al. (1999) Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285: 727-729.
-
Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, et al. (2008) Tumor-associated MICA is shed by ADAM proteases. Cancer Res 68: 6368-6376.
-
Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, et al. (1999) Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci U S A 96: 6879-6884.
-
Groh V, Wu J, Yee C, Spies T (2002) Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 419: 734-738.
-
Poggi A, Zocchi MR (2013) How to exploit stress-related immunity against Hodgkin's lymphoma: Targeting ERp5 and ADAM sheddases. Oncoimmunology 2: e27089.
-
Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, et al. (2007) Disulphide-isomerase-enabled shedding of tumor-associated NKG2D ligands. Nature 447: 482-486.
-
Chitadze G, Lettau M, Bhat J, Wesch D, Steinle A, et al. (2013) Shedding of endogenous MHC class I-related chain molecules A and B (MICA and MICB) from different human tumor entities: Heterogeneous involvement of the a disintegrin and metalloproteases 10 and 17 (ADAM10 and ADAM17). Int J Cancer 133: 1557-1566.
-
Wang X, Lundgren AD, Singh P, Goodlett DR, Plymate SR, et al. (2009) An six-amino acid motif in the alpha3 domain of MICA is the cancer therapeutic target to inhibit shedding. Biochem Biophys Res Commun 387: 476-481.
-
Paschen A, Baingo J, Schadendorf D (2014) Expression of stress ligands of the immunoreceptor NKG2D in melanoma: regulation and clinical significance. Eur J Cell Biol 93: 49-54.
-
Kaifu T, Escalière B, Gastinel LN, Vivier E, Baratin M (2011) B7-H6/NKp30 interaction: a mechanism of alerting NK cells against tumors. Cell Mol Life Sci 68: 3531-3539.
-
Wu MR, Zhang T, Gacerez AT, Coupet TA, DeMars LR, et al. (2015) B7H6-Specific Bispecific T Cell Engagers Lead to Tumor Elimination and Host Antitumor Immunity. J Immunol 194: 5305-5311.
-
Kellner C, Maurer T, Hallack D, Repp R, van de Winkel JG, et al. (2012) Mimicking an induced self phenotype by coating lymphomas with the NKp30 ligand B7-H6 promotes NK cell cytotoxicity. J Immunol 189: 5037-5046.
-
Schlecker E, Fiegler N, Arnold A, Altevogt P, Rose-John S, et al. (2014) Metalloprotease-mediated tumor cell shedding of B7-H6, the ligand of the natural killer cell activating receptor NKp30. Cancer Res 74: 3429-3440.
-
Curtin JF, Cotter TG (2003) Live and let die: regulatory mechanisms in Fas-mediated apoptosis. Cell Signal 15: 983-992.
-
French LE, Tschopp J (2002) Defective death receptor signaling as a cause of tumor immune escape. Semin Cancer Biol 12: 51-55.
-
Chen L, Park SM, Tumanov AV, Hau A, Sawada K, et al. (2010) CD95 promotes tumour growth. Nature 465: 492-496.
-
Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, et al. (2005) Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res 11: 1010-1020.
-
Favre-Felix N, Fromentin A, Hammann A, Solary E, Martin F, et al. (2000) Cutting edge: the tumor counterattack hypothesis revisited: colon cancer cells do not induce T cell apoptosis via the Fas (CD95, APO-1) pathway. J Immunol 164: 5023-5027.
-
Prado-Garcia H, Aguilar-Cazares D, Meneses-Flores M, Morales-Fuentes J, Lopez-Gonzalez JS (2008) Lung carcinomas do not induce T-cell apoptosis via the Fas/Fas ligand pathway but down-regulate CD3 epsilon expression. Cancer Immunol Immunother 57: 325-336.
-
Ebsen H, Lettau M, Kabelitz D, Janssen O (2015) Subcellular localization and activation of ADAM proteases in the context of FasL shedding in T lymphocytes. Mol Immunol 65: 416-428.
-
Fichter CD, Timme S, Braun JA, Gudernatsch V, Schopflin A, et al. (2014) EGFR, HER2 and HER3 dimerization patterns guide targeted inhibition in two histotypes of esophageal cancer. Int J Cancer 135: 1517-1530.
-
Anderson DR, Grillo-López A, Varns C, Chambers KS, Hanna N (1997) Targeted anti-cancer therapy using rituximab, a chimaeric anti-CD20 antibody (IDEC-C2B8) in the treatment of non-Hodgkin's B-cell lymphoma. Biochem Soc Trans 25: 705-708.
-
Romee R, Foley B, Lenvik T, Wang Y, Zhang B, et al. (2013) NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 121: 3599-3608.
-
Wang Y, Wu J, Newton R, Bahaie NS, Long C, et al. (2013) ADAM17 cleaves CD16b (FcγRIIIb) in human neutrophils. Biochim Biophys Acta 1833: 680-685.
-
Kubota T, Niwa R, Satoh M, Akinaga S, Shitara K, et al. (2009) Engineered therapeutic antibodies with improved effector functions. Cancer Sci 100: 1566-1572.
-
Ratner M (2014) Genentech's glyco-engineered antibody to succeed Rituxan. Nat Biotechnol 32: 6-7.
-
Jing Y, Ni Z, Wu J, Higgins L, Markowski TW, et al. (2015) Identification of an ADAM17 cleavage region in human CD16 (FcγRIII) and the engineering of a non-cleavable version of the receptor in NK cells. PLoS One 10: e0121788.
-
Lajoie L, Congy-Jolivet N, Bolzec A, Gouilleux-Gruart V, Sicard E, et al. (2014) ADAM17-mediated shedding of FcγRIIIA on human NK cells: identification of the cleavage site and relationship with activation. J Immunol 192: 741-751.
-
Xin-Ji Z, Yong-Gang L, Xiao-Jun S, Xiao-Wu C, Dong Z, et al. (2015) The prognostic role of neutrophils to lymphocytes ratio and platelet count in gastric cancer: A meta-analysis. Int J Surg 21: 84-91.
-
Albanesi M, Mancardi DA, Jonsson F, Iannascoli B, Fiette L, et al. (2013) Neutrophils mediate antibody-induced antitumor effects in mice. Blood 122: 3160-3164.
-
Tang J, Zarbock A, Gomez I, Wilson CL, Lefort CT, et al. (2011) Adam17-dependent shedding limits early neutrophil influx but does not alter early monocyte recruitment to inflammatory sites. Blood 118: 786-794.
-
Schleiffenbaum B, Spertini O, Tedder TF (1992) Soluble L-selectin is present in human plasma at high levels and retains functional activity. J Cell Biol 119: 229-238.
-
Yamanaka YJ, Gierahn TM, Love JC (2013) The dynamic lives of T cells: new approaches and themes. Trends Immunol 34: 59-66.
-
Baitsch L, Fuertes-Marraco SA, Legat A, Meyer C, Speiser DE (2012) The three main stumbling blocks for anticancer T cells. Trends Immunol 33: 364-372.
-
Pauken KE, Wherry EJ (2015) Overcoming T cell exhaustion in infection and cancer. Trends Immunol 36: 265-276.
-
Li N, Wang Y, Forbes K, Vignali KM, Heale BS, et al. (2007) Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J 26: 494-504.
-
Pinton L, Solito S, Damuzzo V, Francescato S, Pozzuoli A, et al. (2016) Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget 7: 1168-1184.
-
Moller-Hackbarth K, Dewitz C, Schweigert O, Trad A, Garbers C, et al. (2013) A disintegrin and metalloprotease (ADAM) 10 and ADAM17 are major sheddases of T cell immunoglobulin and mucin domain 3 (Tim-3). J Biol Chem 288: 34529-34544.
-
Anderson AC (2014) Tim-3: an emerging target in the cancer immunotherapy landscape. Cancer Immunol Res 2: 393-398.
-
Schweigert O, Dewitz C, Moller-Hackbarth K, Trad A, Garbers C, et al. (2013) Soluble T cell immunoglobulin and mucin domain (TIM-) 1 and 4 generated by A Disintegrin And Metalloproteases (ADAM-)10 and 17 bind to phosphatidylserine. Biochim Biophys Acta 1843: 275-287.
-
Meyers JH, Chakravarti S, Schlesinger D, Illes Z, Waldner H, et al. (2005) TIM-4 is the ligand for TIM-1, and the TIM-1-TIM-4 interaction regulates T cell proliferation. Nat Immunol 6: 455-464.
-
Driscoll WS, Vaisar T, Tang J, Wilson CL, Raines EW (2013) Macrophage ADAM17 deficiency augments CD36-dependent apoptotic cell uptake and the linked anti-inflammatory phenotype. Circ Res 113: 52-61.
-
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646-674.
-
Etzerodt A, Rasmussen MR, Svendsen P, Chalaris A, Schwarz J, et al. (2014) Structural basis for inflammation-driven shedding of CD163 ectodomain and tumor necrosis factor-α in macrophages. J Biol Chem 289: 778-788.
-
Yacoub D, Benslimane N, Al-Zoobi L, Hassan G, Nadiri A, et al. (2013) CD154 is released from T-cells by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and ADAM17 in a CD40 protein-dependent manner. J Biol Chem 288: 36083-36093.
-
Esposito P, Rampino T, Gregorini M, Gabanti E, Bianzina S, et al. (2012) Mechanisms underlying sCD40 production in hemodialysis patients. Cell Immunol 278: 10-15.
-
Zaiss DM, van Loosdregt J, Gorlani A, Bekker CP, Grone A, et al. (2013) Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity 38: 275-284.
-
Yuan CH, Sun XM, Zhu CL, Liu SP, Wu L, et al. (2015) Amphiregulin activates regulatory T lymphocytes and suppresses CD8+ T cell-mediated anti-tumor response in hepatocellular carcinoma cells. Oncotarget 6: 32138-32153.
-
Dai K, Huang L, Sun X, Yang L, Gong Z (2015) Hepatic CD206-positive macrophages express amphiregulin to promote the immunosuppressive activity of regulatory T cells in HBV infection. J Leukoc Biol 98: 1071-1080.
-
Hsu YL, Huang MS, Cheng DE, Hung JY, Yang CJ, et al. (2011) Lung tumor-associated dendritic cell-derived amphiregulin increased cancer progression. J Immunol 187: 1733-1744.
-
Zhou X, Tang J, Cao H, Fan H, et al. (2015) Tissue resident regulatory T cells: novel therapeutic targets for human disease. Cell Mol Immunol 12: 543-552.
-
Carvalho S, Lindzen M, Lauriola M, Shirazi N, Sinha S, et al. (2016) An antibody to amphiregulin, an abundant growth factor in patients' fluids, inhibits ovarian tumors. Oncogene 35: 438-447.
-
Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S (2009) Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J Immunol 183: 937-944.
-
Li Y, Brazzell J, Herrera A, Walcheck B (2006) ADAM17 deficiency by mature neutrophils has differential effects on L-selectin shedding. Blood 108: 2275-2279.
-
Boutet P, Aguera-Gonzalez S, Atkinson S, Pennington CJ, Edwards DR, et al. (2009) Cutting edge: the metalloproteinase ADAM17/TNF-alpha-converting enzyme regulates proteolytic shedding of the MHC class I-related chain B protein. J Immunol 182: 49-53.
