

International Journal of Diabetes and Clinical Research
Does GABA Metabolism Play any Specific Role in the Stimulation of Insulin Secretion in β-Cells?
J. Pizarro-Delgado and J. Tamarit-Rodriguez*
Biochemistry Department, Medical School, Complutense University, Spain
*Corresponding author:
J. Tamarit-Rodriguez, Biochemistry Department, Medical School, Complutense University, Madrid-28040, Spain, Tel: +34-913941449; E-mail: tamarit@ucm.es
Int J Diabetes Clin Res, IJDCR-1-007, (Volume 1, Issue 2), Review Article; ISSN: 2377-3634
Received: October 03, 2014 | Accepted: November 01, 2014 | Published: November 05, 2014
Citation: Pizarro-Delgado J, Tamarit-Rodriguez J (2014) Does GABA Metabolism Play any Specific Role in the Stimulation of Insulin Secretion in β-Cells?. Int J Diabetes Clin Res 1:007. 10.23937/2377-3634/1410007
Copyright: ©2014 Pizarro-Delgado J. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
It was recognized quite a few years ago that islet GABA metabolism might be implicated in the stimulation of insulin secretion by glucose. Since then, some studies have been addressed to investigate how GABA release by β-cells might exert a negative autocrine control of glucose-stimulated insulin secretion and a parallel paracrine inhibition of glucagon release trough the activation of GABAB and GABAA receptors, respectively. More recently, it has been shown that some nutrient secretagogues like glucose and α-ketoisocaproic acid (KIC) suppress islet GABA release simultaneously to the stimulation of insulin secretion. Some research groups have found that the suppression of GABA release might be a consequence of its increased metabolism. This review summarizes the known biochemical steps implicated in the synthesis of GABA in β-cells and in the acceleration of its metabolism by glucose and KIC in the "GABA-shunt". 2-oxoglutarate (2OG) generation in the citric acid cycle during glucose metabolism or in the transamination of KIC with glutamate (Glu) secondarily feeds the GABA-transamination reaction (GABA-T) that results in the production of semialdehyde succinic acid (SSA) and Glu. SSA may be further metabolized in the "GABA-shunt" (SSA-dehydrogenase) to succinic acid that enters in the citric acid cycle for its further oxidation. The 2OG-dehydrogenase is a limiting enzymatic step in the citric acid cycle and it is proposed that 2OG metabolism in the "GABA-shunt" may bypass this restriction allowing a higher maximum rate of glucose oxidation and of insulin release. Inhibition of GABA-T (with 1mM gabaculine) suppresses the stimulation of insulin secretion and the production of ATP due to the mitochondrial metabolism of 2OG, together with ADP and GABA, in islets permeabilized to small substrates and cofactors with 70mM KCl.
GABA Metabolism in β-cells
It has been known for many years that β-cells express the enzyme required for the synthesis of GABA: L-glutamate is decarboxylated to GABA by any of two glutamic acid decarboxylase isoforms (GAD 65 and GAD 67). GAD 65 is found associated to the �synaptic like microvesicles� in β-cells whereas GAD 67 is a soluble enzyme [1,2]. It is known that GAD 65 antibodies are one of the most prevalent markers of β-cell destruction in type-1 diabetes [3]. Islet GABA synthesis is very dependent on the extracellular L-glutamine concentration (Gln), the most abundant amino acid in plasma (0.5 to 1.0mM): at 0.5mM the content of islet GABA reaches already a maximum that remains stable at 5 and 10mM [4]. Therefore, in rat islets, GABA synthesis is probably always almost saturated at the Gln concentrations prevailing in plasma (0.5-1.0mM).
GABA is further metabolized in the mitochondria following the so called �GABA shunt� pathway [5] (Figure 1). Nothing is known about the mitochondrial membrane transporter responsible of the translocation of GABA from the cytosol into the mitochondria. In the mitochondrial matrix, GABA is deaminated by GABA transaminase (GABA-T) to succinic semialdehyde acid (SSA) and its γ-amino group transferred to 2-oxoglutarate (2OG) to generate L-glutamate. The source of 2OG for GABA-T reaction might proceed from the citric acid cycle during glucose metabolism or from the oxidative deamination of Glu by the glutamate dehydrogenase enzyme (GDH) [4,6]. In vitro, saturated with the corresponding substrates, islet GDH equilibrium is significantly displaced in favor of Glu-amination [4]. In support of this observation, increasing the intracellular Glu concentration by extracellular L-glutamine (Gln) or a permeable form of Glu (dimethyl ester derivative) did not increase insulin secretion unless GDH activity was stimulated by L-leucine: it accelerated mainly glutamate amination and, to a lower degree, also the deamination rate that provides 2OG to the citric acid cycle [4]. SSA may enter the citric acid cycle after its oxidation to succinic acid by SSA-dehydrogenase or being reduced to γ-hydroxybutyrate (GHB) by SSA-reductase [5,7].
GABA has been also claimed to exert a negative autocrine and paracrine control of insulin and glucagon secretion, respectively, by acting on specific GABAB [8,9] or GABAA receptors [10] on β and α-cells of pancreatic islets. This is another potentially important functional aspect of β-cell GABA production that is not treated in this review as extensively as it would deserve because the review has been focused on the discussion of traditional metabolic aspects. Anyhow, our previous work has shown that the stimulation of islet insulin secretion by α-ketoisocaproic acid (KIC or oxo-4-methyl pentanoic acid) and glucose is accompanied of a strong suppression of GABA release by rat pancreatic islets [7,11]. Therefore, a potential autocrine inhibition of insulin secretion by a basal or stimulated release of GABA might be counteracted by glucose or KIC.
Promotion of Islet GABA Catabolism by Nutrient Secretagogues
It has been demonstrated that high glucose decreases both the islet content and release of GABA and concluded that it is the consequence of an increased γ-amine catabolism: both D-mannoheptulose (a competitive glucokinase inhibitor) and gabaculine (a specific GABA-T inhibitor) counteracted glucose induced decrease of islet GABA [11-13]. In fact, a positive linear correlation was found between GABA release and content at different Gln concentrations [11].
KIC also diminished very strongly islet GABA content through an increase of its endogenous catabolism [7,14]: KIC is transaminated to Leu by the branched-chain amino acid transferase (BCAT) and the resulting α-amino acid accumulates extracellularly; in parallel, Glu is deaminated to 2OG [Fig. 1]. In a subsequent transamination reaction, the resulting 2OG is transaminated with GABA (GABA-T reaction) to generate Glu and semialdehyde succinic acid. 4-Methylvaleric acid, an inhibitor of BCAT, suppressed the transamination of KIC [14] and gabaculine reversed KIC-induced decrease of islet GABA content [7]. Similar results were obtained with α-ketohexanoic acid (KC, the α-ketoacid derivative of nor-leucine). In summary, the generation of 2OG in the citric acid cycle by either glucose or KIC seems to promote the metabolism of islet GABA. In fact, a membrane permeable form of 2OG (the dimethylester) dose-dependently increased islet Glu content but decreased GABA content at high concentrations [4].
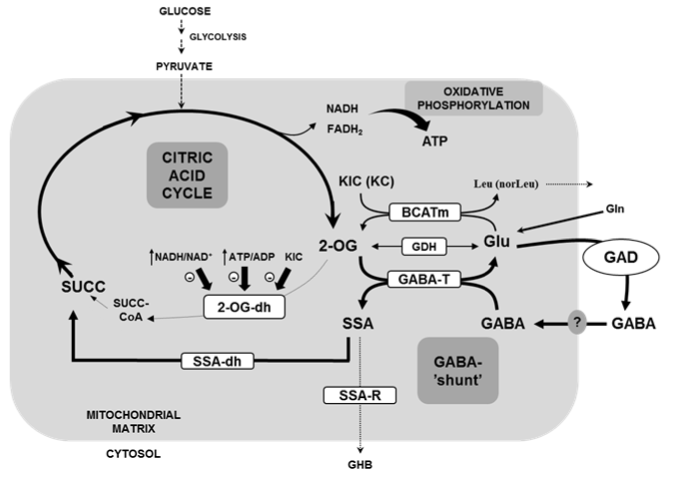
Figure 1: Scheme of the metabolic interconnections between the citric acid
cycle and the �GABA-shunt�
BCATm: branched-chain amino acid transferase, mitochondrial isozyme; 2OG:
2-oxoglutarate or α-ketoglutarate; 2OGdh: 2-oxoglutarate dehydrogenase;
GDH: Glutamic acid dehydrogenase; GAD: Glutamic acid decarboxylase;
GABA-T: GABA transaminase; SSA-dh: Semialdehyde succinic acid
dehydrogenase; SSA-R: Semialdehyde succinic acid reductase; KIC: oxo-4-
methylpentanoate or α-ketoisocaproate; KC: oxohexanoate or a-ketocaproate;
GHB: γ-hydroxybutyrate
View Figure 1
Islet GABA catabolism contributes to the stimulation of insulin secretion
The insulin secretory response of rat perifused islets to 20mM glucose is significantly suppressed by 0.25mM gabaculine (a specific GABA-T inhibitor) at different Gln concentrations. The drug also decreases glucose-induced elevation of islet ATP content and the ATP/ADP ratio at 1mM [11]. Succinic semialdehyde acid (SSA) turned out to be permeable through β-cell plasma membrane and stimulated, similarly to glucose, a biphasic insulin secretion [11]. The second phase of secretion was diminished by 1mM gabaculine at different Gln concentrations. By reversion of the GABA-T reaction, SSA generated an increased content of GABA and one might conclude that its metabolism both in the citric acid cycle (through its conversion to succinic acid) and in the �GABA-shunt� contribute to the measured elevation of islet ATP and ATP/ADP ratio [11].
The biphasic secretory response elicited by any of the used branched-chain 2-oxoacids (BCKA) was strongly suppressed by a BCAT inhibitor [14]: 5mM gabapentin, more specific of the cytosolic isoform, decreased within more than 50% the second phase of stimulation of insulin secretion by oxo-3-methylpentanoate (KMV or α-keto-β-methyl valeric acid, the corresponding keto form of valine). 4-Methylvaleric acid, a general inhibitor of both the cytosolic and mitochondrial BCAT isoforms, suppressed by more than 90% the secretory response of incubated rat islets to KIC. BCKA stimulation of insulin secretion was also dependent on the promotion of endogenous GABA metabolism: the secretory response to KIC in perifused rat islets was suppressed within 26% by 250�M gabaculine [7]. Moreover, depletion of islet GABA content by depolarization with 70mM KCl decreased the second phase of insulin secretion triggered by either KIC or KC within more than 60% [14].
Mechanism of the Contribution of �GABA-shunt� Catabolism to the Stimulation of Glucose-Induced Insulin Secretion
We proposed the hypothesis that the "GABA-shunt" is necessary to bypass a restriction of the metabolic flow in the citric acid cycle imposed by a relatively low 2OG-dehydrogenase activity or expression in β-cells. It has been shown in rat islets that 2OG-dehydrogenase activity is several fold lower that GABA-T activity and both enzymes show similar Km-values to their common substrate 2OG [12,13]. At the mRNA level, 2OG-dehydrogenase enzyme was the least expressed among the six genes studied in rat islets: steady mRNA levels of GAD65, GABA-T and succinic acid dehydrogenase were several-fold higher than 2OG-dehydrogenase mRNA [11].
Recent experiments have made possible to demonstrate that 2OG-driven mitochondrial ATP production and insulin secretion are dependent on the contribution of GABA-T activity using rat islets �permeabilized� with 70mM KCl [17]. This islet preparation has been shown to respond to increases in the extracellular ATP concentration with increments in the biphasic stimulation of insulin secretion [17]. Depolarization with 70mM KCl makes islet β-cells permeable to small metabolites and cofactors bellow an exclusion limit of 1KD due to the opening of connexin36 hemichannels at the plasma membrane of β-cells. This model has allowed us to measure the amount of ATP produced and the rate of insulin released in permeabilized islets incubated or perifused with 5mM 2OG and 5mM ADP in the presence of 2mM GABA. 2OG metabolism in the mitochondria of permeabilized cells promotes the phosphorylation of the accompanying ADP and the resulting ATP equilibrates between the cytosol and extracellular medium were it can be measured. The permeabilized islets are capable to respond reversibly with an increased rate of insulin secretion to an increase concentration of metabolites in the perifusate.
As shown in Figure 2, islet depolarization with 70mM KCl induced a transient, monophasic release of insulin in perifused rat islets that faded out after approximately 10 min. When 5mM 2OG, together with 5mM ADP and 2mM GABA were simultaneously perifused (mins. 15 to 55), the height of the first peak of secretion due to KCl depolarization was not modified but a second phase was triggered and maintained at half the rate recorded at the first peak until the end of the stimulation. Insulin release returned fast to basal values after withdrawal of the metabolic stimulus. The simultaneous presence of 1mM gabaculine (a GABA-T inhibitor), suppressed the second phase of release stimulated by 2OG within almost 50% (Figure 2).
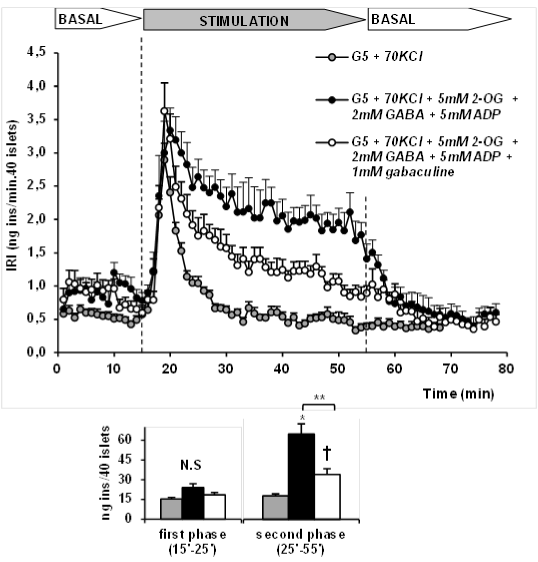
Figure 2:Stimulation of insulin secretion by 5mM 2-oxoglutarate (2OG) in the
presence of 5mM ADP and 2mM GABA in perifused rat islets depolarized with
70mM KCl. Effect of GABA-T inhibition with 1mM gabaculine.
Groups of 40 rat islets were perifused at 5mM glucose (G5, BASAL) for 30
min, then depolarized with 70mM KCl (n=17; grey circles) alone or together
with 5mM 2OG + 5mM ADP + 2mM GABA in the absence (n=8, black
circles) and presence of 1mM gabaculine (n=10, white circles) for another
40 min (STIMULATION) and finally switched back to G5 alone for the last
20min (BASAL again). Insulin concentration was measured at 1 min intervals
in the perifusate after minute 15th. Addition of extra KCl was compensated
by decreasing an equimolar amount of NaCl. The histograms below the
perifusions' panel show the statistical comparisons between the total amount of
insulin secreted under the two different experimental conditions during the first
(15-25min) and second (25-55 min) phase of stimulation. Insulin concentration
in the perifusate was radioimmunologically measured every minute using rat
insulin as a standard (see reference 17 for a more detailed description of
the method). (*p< 0.0001, and �p< 0.001 compared with the corresponding bar
to the left in the absence of perifusate metabolites; **p< 0.002 between the
indicated bars; n=number of experiments)
View Figure 2
The amount of ATP produced and accumulated in the medium after 1 h of incubation of KCl-permeabilized islets, as well as the islet ATP content and the corresponding ATP/ADP ratio were measured with the luciferin/luciferase system. Figure 3 shows that a nanomolar amount of ATP accumulated in the medium after incubation with 5mM 2OG in presence of 5mM ADP and 2mM GABA and it was suppressed within almost 40 % by the additional presence of 1mM gabaculine. The islet content of ATP (3.96 � 0.46, n=8, vs. 4.78 � 0.50 pmol/islet, n=8; N.S.), as well as the corresponding ATP/ADP ratio (1.51 � 0.16, n=8, vs. 1.72 � 0.25, n=8; N.S.), showed a trend to be decreased by 1mM gabaculine.
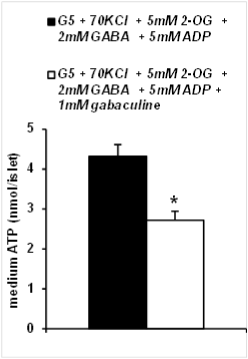
Figure 3: ATP production by rat islets permeabilized with 70mM KCl and
stimulated by 5mM 2-oxoglutarate (2OG) in the presence of 5mM ADP and
2mM GABA. Effect of GABA-T inhibition with 1mM gabaculine
Three groups, each of 25 islets, were incubated at 37�C for 60 minutes in
25�l of Krebs Ringer bicarbonate buffered with 2mM HEPES. The incubation
was stopped on acetone chilled with dry ice and perchloric acid. ATP and
ADP were measured in the supernatant after centrifugation with the luciferin/
luciferase system. (See reference 17 for a detailed description of the method;
*p< 0.005 compared with the corresponding control bar to the left; number of
experiments=9)
View Figure 3
Conclusions
The hypothesis that 2OG-dehydrogenase sets a limit to the metabolic flow in the citric acid cycle that is relieved by the �GABA-shunt� seems to be partially supported by the presented experimental data. Moreover, 2OG-dehydrogenase is subjected to regulation by the NADH/NAD+ and ATP/ADP ratios [12] and has long been considered as a rate-limiting step in the citric acid cycle [18]. Inhibition of 2OG-dehydrogenase by an increased ATP/ADP ratio at high glucose concentrations would subsequently increase the availability of 2OG for GABA-T activity diverting the metabolic flow away from the citric acid cycle into the GABA-shunt. It cannot be discarded that metabolism in the "GABA-shunt" might set the upper limit of islet mitochondrial oxidation and insulin secretion (at least of the second, sustained phase) in response to glucose and other nutrient secretagogues. In the case of stimulation by KIC, 2OG-dehydrogenase activity would not only be restricted by an elevated ATP/ADP ratio but by a direct inhibition of the enzyme activity by KIC itself [19].
References
-
Michalik M, Nelson J, Erecinska M (1993) GABA production in rat islets of Langerhans. Diabetes 42: 1506-1513.
-
Shi Y, Kanaani J, Menard-Rose V, Ma YH, Chang PY, et al. (2000) Increased expression of GAD65 and GABA in pancreatic β-cells impairs first-phase insulin secretion. Am J Physiol Endocrinol Metab 279: E684-E694.
-
Degli Esposti M, Mackay IR (1997) The GABA network and the pathogenesis of IDDM. Diabetologia 40: 352-356.
-
Fern�ndez-Pascual S, Mukala-Nsengu-Tshibangu A, Mart�n Del R�o R, Tamarit-Rodr�guez J (2004) Conversion into GABA (gamma-aminobutyric acid) may reduce the capacity of L-glutamine as an insulin secretagogue. Biochem J 379: 721-729.
-
Sorenson RL, Garry DG, Brelje TC (1991) Structural and functional considerations of GABA in islets of Langerhans. Beta-cells and nerves. Diabetes 40: 1365-1374.
-
Fahien LA, Macdonald MJ (2011) The complex mechanism of glutamate dehydrogenase in insulin secretion. Diabetes 60: 2450-2454.
-
Hern�ndez-Fisac I, Fern�ndez-Pascual S, Orts�ter H, Pizarro-Delgado J, Mart�n del R�o R, et al. (2006) Oxo-4-methylpentanoic acid directs the metabolism of GABA into the citric acid cycle in rat pancreatic islets. Biochem J 400: 81-89.
-
Brice NL, Varadi A, Ashcroft SJ, Molnar E (2002) Metabotropic glutamate and GABA(B) receptors contribute to the modulation of glucose-stimulated insulin secretion in pancreatic beta cells. Diabetologia 45: 242-252.
-
Braun M, Wendt A, Buschard K, Salehi A, Sewing S, et al. (2004) GABAB receptor activation inhibits exocytosis in rat pancreatic β-cells by G-protein-dependent activation of calcineurin. J Physiol 559: 397-409.
-
Wendt A, Birnir B, Buschard K, Gromada J, Salehi A, et al. (2004) Glucose inhibition of glucagon secretion from rat a-cells is mediated by GABA release from neighboring cells. Diabetes 53: 1038-1045.
-
Pizarro-Delgado J, Braun M, Hern�ndez-Fisac I, Mart�n-Del-R�o R, Tamarit-Rodriguez J (2010) Glucose promotion of GABA metabolism contributes to the stimulation of insulin secretion in β-cells. Biochem J 431: 381-389.
-
Winnock F, Ling Z, De Proft R, Dejonghe S, Schuit F, et al. (2002) Correlation between GABA release from rat islet beta-cells and their metabolic state. Am J Physiol Endocrinol Metab 282: E937-942.
-
Wang C, Kerckhofs K, Van de Casteele M, Smolders I, Pipeleers D, et al. (2006) Glucose inhibits GABA release by pancreatic beta-cells through an increase in GABA shunt activity. Am J Physiol Endocrinol Metab 290: E494-499.
-
Pizarro-Delgado J, Hern�ndez-Fisac I, Mart�n-Del-R�o R, Tamarit-Rodriguez J (2009) Branched-chain 2-oxoacids transamination increases �GABA-shunt� metabolism and insulin secretion in isolated islets. Biochem J 419: 359-368. q
-
Heckert LL, Butler MH, Reimers JM, Albe KR, Wright BE (1989) Purification and characterization of the 2-oxoglutarate dehydrogenase complex from Dictyostelium discoideum. J Gen Microbiol 135: 155-161.
-
Cash C, Maitre M, Ciesielski L, Mandel P (1974) Purification and partial characterisation of 4-aminobutyrate 2-ketoglutarate transaminase from human brain. FEBS Lett 47: 199-203.
-
Pizarro-Delgado J, Fasciani I, Temperan A, Romero M, Gonz�lez-Nieto D, et al. (2014) Inhibition of connexin36 hemichannels by glucose contributes to the stimulation of insulin secretion. Am J Physiol Endocrinol Metab 306: E1354-E1366.
-
Roche TE, Lawlis VB (1982) Structure and regulation of alpha-ketoglutarate dehydrogenase of bovine kidney. Ann N Y Acad Sci 378: 236-249.
-
Patel MS (1974) Inhibition by the branched-chain 2-oxo acids of the 2-oxoglutarate dehydrogenase complex in developing rat and human brain. Biochem J 144: 91-97.
