

International Journal of Diabetes and Clinical Research
Torpid Diabetic Wound Healing: Evidence on the Role of Epigenetic Forces
Jorge Berlanga-Acosta1*, Yssel Mendoza-Mari1, Maday Fernandez-Mayola1, Ariana Garcia-Ojalvo1, Calixto Valdes-Perez2, William Savigne-Gutierrez2, Daniel Reynaldo-Concepcion2, Ileydis Iglesias-Marichal3, angel Abreu-Cruz4, Celso Suarez-Lescay5, Ana Mir-Benitez6, Natacha Sancho-Soutelo7 and Luis Herrera-Martinez1
1Tissue Repair and Cyto-protection Research Group, Center for Genetic Engineering and Biotechnology, Cuba
2Diabetic Angiopathy Ward, National Institute of Angiology and Vascular Surgery, Cuba
3Center for Integral Care of Diabetic Patients, Cuba
4Center for Surgical and Medical Research, Cuba
5Diabetic Angiopathy Service, Hospital Militar Joaquin Castillo Duany, Cuba
6Department of Plastic Surgery, Hospital Joaquin Albarran, Cuba
7Hospital Juan Bruno Zayas, Carretera del Caney, Reparto Pastorita, Cuba
*Corresponding author:
Jorge Berlanga-Acosta, Tissue Repair and Cyto-protection Research Group, Center for Genetic Engineering and Biotechnology, Playa. PO Box: 10600 Havana, Cuba, E-mail: jorge.berlanga@cigb.edu.cu
Int J Diabetes Clin Res, IJDCR-2-020, (Volume 2, Issue 1), Research Article; ISSN: 2377-3634
Received: October 23, 2014 | Accepted: January 26, 2015 | Published: January 29, 2015
Citation: Berlanga-Acosta J, Mendoza-Mari Y, Fernandez-Mayola M, Garcia-Ojalvo A, Valdes-Perez C, et al., (2015) Torpid Diabetic Wound Healing: Evidence on the Role of Epigenetic Forces. Int J Diabetes Clin Res 2:020. 10.23937/2377-3634/1410020
Copyright: © 2015 Berlanga-Acosta J, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The increasing number of diabetes patients represents a health challenge due to disease-related, end-organs complications. Hyperglycemia is considered the proximal trigger of an intricate cascade of molecular processes that progressively deteriorate tissues and organs, leading to the onset of clinical complications. Lower extremity ulcerations and their ensued refractoriness to heal can potentially result in amputation and disability and remain the second most feared diabetic complication. We have identified particular morphogenetic traits in diabetic foot ulcer granulation tissues and its cultured fibroblasts. Diabetic ulcer-derived fibroblasts conserve a sort of memory as their in vitro traits very much recapitulate the in vivo behavior, in terms of proliferative disabilities and transcriptional and post-translational modifications of genes involved in proliferation, migration and ECM dinamics. Furthermore, the acute, in vivo morphologic recreation of a microangiopathy in a neo-formed granulation tissue is worth mentioning. All these elements suggest that �metabolic memory,� in which chromatin remodeling and long-lasting epigenetic changes play important roles, could contribute to the persistence of diabetic complications. Metabolic memory is largely responsible for the onset/perpetuation of the ulcers chronicity phenotype . The comprehensive understanding of the chromatin choreography underlying this pathogenic stream; and its potential pharmacologic manipulation would allow for future innovative therapies for diabetic complications, including wound healing refractoriness.
Keywords
Diabetes, Diabetic foot ulcers, Chronic wounds, Metabolic memory, Epigenetics
Abbreviations
T2DM: Type 2-diabetes mellitus, AGEs: Advanced Glycation-End Products, ROS: Reactive Oxygen Species, NFkB p65: Nuclear Factor-Kappa B, PIK3CA: Phosphatidylinositol-4,5-Bisphosphate 3-Kinase, MTOR: Mammalian Target of Rapamycin, FOXO: Forkhead Box O, RT-PCR: Reverse Transcription-Polymerase Chain Reaction
Introduction
Type 2-diabetes mellitus (T2DM) is a group of metabolic disorders that is currently expanding in a pandemic magnitude [1].
Hyperglycemia has been defined by the World Health Organization as a condition in which fasting blood glucose levels are greater than 7.0mmol/L (126 mg/dL) or greater than 11.0 mmol/L (200 mg/dL), 2 hours after meals [2]. Sustained hyperglycemic state is invoked as the proximal trigger for diabetes-associated biochemical disturbances and the ensued end-organs complications [3] Hyperglycemic condition is associated with systemic endothelial dysfunction, a central factor for the onset and progression of macro and microvascular complications which eventually undermine whole organ systems [4].
Although building the molecular bridge between the trivial episodes of hyperglycemia and gene transcription machinery still stands as a challenge, mounting evidence sustain the pathogenic role of epigenetic mechanisms in hyperglycemia-induced organs complications. The experimental and clinical evidences which support the concept of �metabolic memory�, as the cellular ability to remember hyperglycemic experiences and thus perpetuate diabetic complications even under normal blood glucose levels [5,6], have offered explanation for the defective wound healing traits of diabetic ulcer cells.
Chronic wounds are commonly defined as wounds that do not follow the well-defined stepwise process of physiological healing but are trapped in an uncoordinated and self-perpetuated phase of inflammation. As a result, the healing process is delayed, incomplete, and/or asynchronous; thus resulting in poor anatomical and functional outcomes [7]. Among chronic wounds, lower extremities ulceration is outlined as one of the most complex to treat. Diabetic foot ulcer, with the subsequent healing failure, is associated with amputation-disability, morbidity and mortality [8]. Irrespective to the research and financial efforts put forward for years, current figures of amputations among the diabetic population are alarming since every 20 seconds some individual is amputated [9].
There is no single, universal mechanism to explain why cutaneous wounds fail to heal in diabetic subjects. Rather, it is a multifactorial event so that diverse cellular and humoral factors interact in disrupting more than one of the phases of the healing process. Aside from the local and systemic functional, predisposing factors [10], the evolution to chronicity appears to be influenced by protracted inflammation [11]. Besides, toxic effects induced by the dermal accumulation of Advanced Glycation-End Products (AGEs), Reactive Oxygen Species (ROS) overproduction and an actively recurrent biofilm are also responsible of this hard-to-heal phenotype [11,12].
Here we provide a brief overview and authors considerations about particular experimental observations, which can only be explained by virtue of the cellular ability to retain past metabolic experiences. However, these views and thoughts render further substantiation to this controversial concept.
Wound Healing Phases and Epigenetic
Most chronic wounds show similar behavior and evolution despite etiological differences, which indicates that their development is a heterogeneous and multifactorial process [13]. Stagnancy in granulation, failure of contraction, and delayed re-epithelialization are clinical hallmarks of diabetic lower extremities wounds. Although it is known that multiple driving forces disrupt the cutaneous repair machinery in diabetes-affected individuals, it is intriguing to notice that granulation tissue exhibits malformed vessels that recreate the typical long term diabetic microvascular damages [14]. The question is: what are the mechanisms operating for these vascular morphologic abnormalities, occurring even in a young granulation tissue from a diabetic patient with controlled glucose levels?
Growing data fuel the concept that epigenetic changes are instrumental players for the diabetics" wound healing failure. The inflammatory process in diabetic ulcers is more a condition than a physiological reaction. Pro- inflammatory cytokines reduce fibroblasts and vascular progenitor cells migration, anchorage, activation, and most importantly �entice� these cells to commit suicide [15]. Via nuclear factor-kappaB (NFκB p65) and c-Jun N-terminal kinases signaling pathways [16], inflammation also disrupts extracellular matrix synthesis not only by up-regulating matrix proteases [17] but also by dismantling anabolic pathways usually activated by the agonistic occupation of tyrosine kinase receptors, (including insulin receptor) which at the end, would turn to activate PIK3CA/AKT1-MTOR axis [18]. Under conditions of poor growth factors availability and consequently reduced Akt activity, FOXO is activated and retained in the nuclear compartment, hence shutting down MTOR anabolic activities [19]. The contributions by El-Osta"s laboratory are pivotal to understand the perpetuation of inflammation in diabetes. They demonstrated that NFκB-p65 transcriptional up-regulation resulted from epigenetic marks which modified the nature of the histone methylation on the gene promoter region [20].
Furthermore, particular epigenetic changes have shown to impact on the angiogenesis, and re-epithelialization processes of diabetic models as excellently reviewed by Rafehi and co-workers [21]. The reviewed studies converge to implicate an epigenetic-based metabolic memory in the onset of the phenotype that characterizes diabetic wounds. Thus, it could be attractive to dissect out what the possible contribution could be of ischemia and neuropathy, for the shaping of a specific chromatin remodeling and its input into the ulcer phenotype. Cumulative epigenetic modifications appear to be the driving force en route for the point-of-no return in end-organs complications or simply to irreversible insulin resistance [22].
Cells Involved in Cutaneous Healing Recall their Metabolic Experiences
Acute exposure to high glucose concentrations exerts a detrimental metabolic and bioenergetic effect on cutaneous fibroblasts [23]. Early observations suggested the existence of an intrinsic or imprinted behavioral pattern on cutaneous fibroblasts, since replication did not appear solely impaired in cells harvested from diabetics, but also in cells from diabetes-genetically predisposed subjects [24]. Although the study by Engerman and Kern in 1987 [25] was seminal to shape the future concept of metabolic memory relevant to cutaneous wound healing, the first evidence supporting the metabolic memory concept, emerged from the classic experiment developed by Vracko and Benditt. In 1975, they showed that diabetic patient-derived cells exhibited about half the number of population doublings as compared to cells from non-diabetic donors, indicating a reduced replicative lifespan for diabetic fibroblasts, even under normoglycemic culture conditions [26]. This and other subsequent findings based on cultured fibroblasts suggested that explanted cells from diabetic patients conserve a memory since their in vitro traits recapitulate their in vivo behavior. It was perhaps the primary intuition on the existence of a sort of genetic or epigenetic predisposition for a certain trait, even when the cells no longer remained in the diseased organism.
As mentioned before, only by virtue of the existence of an epigenetic-mediated mechanism it is possible to explain the proliferative refractoriness and the propensity to culture senescence of diabetic ulcers-derived fibroblasts. Analogous to fibroblasts [26], endothelial cells also preserve the imprinting imposed by high glucose burden as they exhibit elevated transcriptional expression rates for both fibronectin and collagen IV even after been switched to normal glucose environment [27]. As a matter of fact the authors of this study were those who coined the term of metabolic memory in 1990.
Molecules and Epigenetics
Animal studies have illustrated the responsibility of the oxidative and nytrosilative stress as instrumentals for the onset of the point-of-no return [28]. In line with these observations is the fact that the diabetic wound is a rich source of oxygen reactive species which generates a intensely toxic microenvironment for fibroblasts and endothelial cells [29]. However, it remains to be answered what could be the echoes of the excess of free radicals for local and peripheral cutaneous cells once they were exposed. In situ ulcer recurrence documented upon short follow-up periods [30] represents a serious problem that entails frustration for the patient and the clinician. Considering the above described evidences it is plausible to hypothesize that ulcer recurrence, irrespective to other predisposing factors, could be a clinical expression form of the point-of-no return. In other words, the early metabolic stress is "remembered" by the cutaneous cells and translated into a permanent, progressive and perpetuated harmful imprinting [31].
The fact that mitochondrial ROS overproduction was identified as proximal in the pathogenic cascade of diabetes complications [32] paved the way for the concept that ROS operates as the putative link between glycemia and the cells" chromatin structure. Mitochondrial excessive superoxide with the ensued ROS spillover stood as the pathogenic core of the �hyperglycemic memory� in which chromatin remodeling and long-lasting epigenetic changes ensure the persistence of inflammation and other molecular disorders involved in diabetes complications. Figure 1 is a diagrammatic representation of the major actors performing in the cascade.
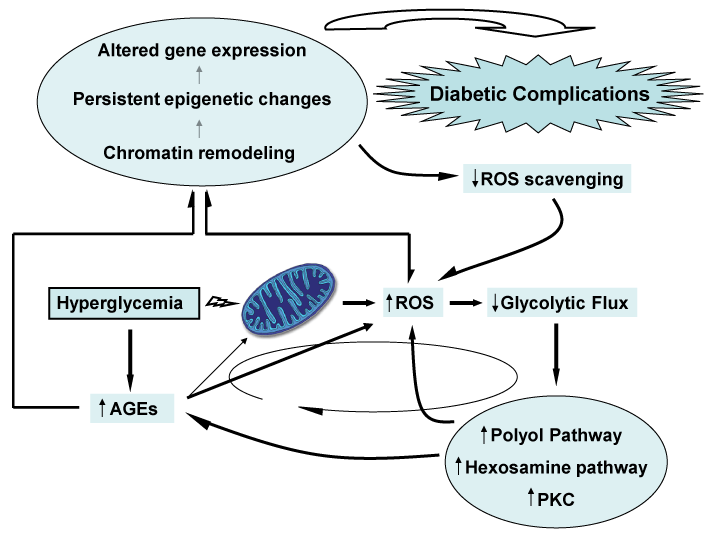
Figure 1: Hyperglycemia strikes mitochondrial functions leading to an
auto-perpetual everlasting metabolic disorder known as, �hyperglycemic
metabolic memory�. Hyperglycemia increases AGEs production thus leading
to the consequent direct ROS generation via AGE/RAGE interaction;
but also via AGEs generation from mitochondrial proteins involved in
oxidative phosphorylation. Increased oxidative stress as a consequence of
mitochondrial ROS generation diminish glycolytic flux by inhibiting GADPH,
thus causing a stimulation of the polyol and hexosamine pathways together
with augmented PKC expression which pivots in AGEs and ROS generation in
a continuous vicious cycle represented with an elliptical arrow. This perpetual
signaling network is supported by persistent epigenetic changes caused by
ROS and AGEs chromatin remodeling through histones modification thus
leading to altered gene expression.
View Figure 1
Our Experience in Metabolic Memory
In an attempt to understand the molecular basis of diabetic wound healing refractoriness, our group has systematically cultured primary granulation tissue fibroblasts from ischemic and neuropathic ulcers. We have confirmed the reduced replicative potential, as compared to age-matched cells from non-diabetic, burn-injured donors. Again, the improvement of culture microenvironment, including adequate oxygen availability and physiological glucose levels, does not ameliorate the proliferative arrest. We observed that this phenotype of arrest appears associated to the overexpression of activated forms of TP53 and CDKN1A (p21) along with a downregulation of the AKT1/MTOR/CCND1 axis [33]. We still miss to learn how long could last these post-translational modifications in those biopsy-derived cells following culture passages.
Recent observations from our group deserve special comment. Not only because they are unprecedented, but particularly because looking through the prism of transmissible epigenetic events, can they be explained [14]. (I) Through the systematic histological analysis of the granulation tissue biopsies from diabetic ischemic and neuropathic ulcers, we distinguished that the ulcer�s major ethiopathogenic component imposes a particular histological pattern of granulation tissue, which is largely similar and privative for ischemics and for neuropathics classes. (II) Microvascular damages as the fibro-hyaline and proliferative arteriolar sclerosis, ordinarily of long term evolution, are found and completely recreated in neo-formed vessels within granulation tissues no older than two weeks. These observations incite to speculate that an aberrant driving force imposes over and impinge the organizational process during fibroangiogenesis. (III) Following comparative RT-PCR studies using clinical biopsies of granulation tissue from pressure ulcers, and diabetics" ischemic and neuropathic ulcers; we detected a significant derangement in a group of well characterized glucose-metabolism related genes in the diabetic ulcers [14]. Diabetic ulcer cells express far less insulin receptor, hexokinase (isoforms 1 and 2), phosphofructokinase, pyruvate kinase (isoforms 1 and 2), pyruvate dehydrogenase, and significantly more of its inhibitor enzyme pyruvate dehydrogenase kinase (isoform 4). We see with interest that granulation tissue, which can be considered as a transient organ made up by �de novo� cells, reproduces the same transcriptional profile of those genes considered as insulin resistance/glucose intolerance markers, and predictors for type-2 diabetes onset, in the liver, skeletal muscle, and adipose tissue as has been broadly described [34-36]. This raises the questions about if the granulation tissue is an additional insulin-resistant organ.
Experimental evidences supporting the relevance of the metabolic memory in tissue repair has extended to adult zebrafish. This fish has been reliably used as a wound healing model through caudal fin regeneration, under normal or diabetic circumstances based on the fin amputation [37]. Since the zebrafish is capable to regenerate its damaged pancreas and restore a euglycemic state similar to what would be expected in post-transplant human patients; multiple rounds of caudal fin amputation allow for the separation and study of pure epigenetic effects. Although euglycemia is achieved following pancreatic regeneration the impaired fin regeneration is retained even after multiple rounds of regeneration in the daughter fin tissues. These elegant experiments conducted in a small aquatic organism converge to support in vitro, as from rodents and clinical evidences of an underlying epigenetic process based on a wrong metabolic experience [38]. The later was subsequently confirmed through microarray and bioinformatic studies demonstrating the aberrant expression of 71 key regulatory genes involved in tissue repair in the diabetic state [39,40]. Above all, the growing zebrafish experiences have left no room to doubt that metabolic memory is a phenomenon broadly represented in animal species.
Concluding Remarks
Diabetes is an exclusive disease that imposes a variety of distal organs complications as some unmatched traits in some cell types of the affected patients. Epigenetic as the resultant governor of cell"s phenotype upon its genome interaction with the environment, has provided an innovative research field and set out an era of hopes for the control of diabetic complications, including an effective and lasting wound healing. Now we stand before the challenge to fully understand the phenomenon since myriad of questions still remain to be answered and controversies continue to exist. As we have learned during the last 20 years that benefits are not the best if glucose control is delayed; it is likely that over the next 20 years we could teach the cells to keep a healthier metabolic memory as to preserve the native chromatin structure.
References
-
Shaw JE, Sicree RA, Zimmet PZ (2010) Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 87: 4-14.
-
American Diabetes Association (2014) Diagnosis and classification of diabetes mellitus. Diabetes Care 37: S81-90.
-
Yan LJ (2014) Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J Diabetes Res 2014: 137919.
-
Patel H, Chen J, Das KC, Kavdia M (2013) Hyperglycemia induces differential change in oxidative stress at gene expression and functional levels in HUVEC and HMVEC. Cardiovasc Diabetol 12: 142.
-
Ceriello A (2012) The emerging challenge in diabetes: the "metabolic memory". Vascul Pharmacol 57: 133-138.
-
El-Osta A (2012) Redox mediating epigenetic changes confer metabolic memories. Circ Res 111: 262-264.
-
(1999) Consensus development conference on diabetic foot wound care. 7-8 April 1999, Boston, MA. American Diabetes Association Adv Wound Care 12: 353-361.
-
McEwen LN, Ylitalo KR, Herman WH, Wrobel JS (2013) Prevalence and risk factors for diabetes-related foot complications in Translating Research Into Action for Diabetes (TRIAD). J Diabetes Complications 27: 588-592.
-
Boulton AJ, Vileikyte L, Ragnarson-Tennvall G, Apelqvist J (2005) The global burden of diabetic foot disease. Lancet 366: 1719-1724.
-
Alavi A, Sibbald RG, Mayer D, Goodman L, Botros M, et al. (2014) Diabetic foot ulcers: Part I. Pathophysiology and prevention. J Am Acad Dermatol 70: e1-18.
-
Bruhn-Olszewska B, Korzon-Burakowska A, Gabig-Cimińska M, Olszewski P, Węgrzyn A, et al. (2012) Molecular factors involved in the development of diabetic foot syndrome. Acta Biochim Pol 59: 507-513.
-
Lipsky BA, Richard JL, Lavigne JP (2013) Diabetic foot ulcer microbiome: one small step for molecular microbiology . . . One giant leap for understanding diabetic foot ulcers? Diabetes 62: 679-681.
-
Blakytny R, Jude E (2006) The molecular biology of chronic wounds and delayed healing in diabetes. Diabet Med 23: 594-608.
-
Mendoza Y, Valdes C, Rodriguez E, Suarez J, Garcia A, et al. (2013) Histological and trancriptional expression differences between Diabetic Foot and Pressure Ulcers. Journal of Diabetes & Metabolism 4: 296.
-
Xu F, Zhang C, Graves DT (2013) Abnormal cell responses and role of TNF-α in impaired diabetic wound healing. Biomed Res Int 2013: 754802.
-
Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, et al. (2004) A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol 6: 97-105.
-
Tecilazich F, Dinh TL, Veves A (2013) Emerging drugs for the treatment of diabetic ulcers. Expert Opin Emerg Drugs 18: 207-217.
-
Xie J, Proud CG (2013) Crosstalk between mTOR complexes. Nat Cell Biol 15: 1263-1265.
-
Hay N1 (2011) Interplay between FOXO, TOR, and Akt. Biochim Biophys Acta 1813: 1965-1970.
-
Brasacchio D, Okabe J, Tikellis C, Balcerczyk A, George P, et al. (2009) Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 58: 1229-1236.
-
Rafehi H, El-Osta A, Karagiannis TC (2011) Genetic and epigenetic events in diabetic wound healing. Int Wound J 8: 12-21.
-
Zhao J, Goldberg J, Bremner JD, Vaccarino V (2012) Global DNA methylation is associated with insulin resistance: a monozygotic twin study. Diabetes 61: 542-546.
-
Dymkowska D, Drabarek B2, Podszywałow-Bartnicka P2, Szczepanowska J2, Zabłocki K2 (2014) Hyperglycaemia modifies energy metabolism and reactive oxygen species formation in endothelial cells in vitro. Arch Biochem Biophys 542: 7-13.
-
Goldstein S, Moerman EJ, Soeldner JS, Gleason RE, Barnett DM (1979) Diabetes mellitus and genetic prediabetes. Decreased replicative capacity of cultured skin fibroblasts. J Clin Invest 63: 358-370.
-
Engerman RL, Kern TS (1987) Progression of incipient diabetic retinopathy during good glycemic control. Diabetes 36: 808-812.
-
Vracko R, Benditt EP (1975) Restricted replicative life-span of diabetic fibroblasts in vitro: its relation to microangiopathy. Fed Proc 34: 68-70.
-
Roy S, Sala R, Cagliero E, Lorenzi M (1990) Overexpression of fibronectin induced by diabetes or high glucose: phenomenon with a memory. Proc Natl Acad Sci U S A 87: 404-408.
-
Kowluru RA, Kanwar M, Kennedy A (2007) Metabolic memory phenomenon and accumulation of peroxynitrite in retinal capillaries. Exp Diabetes Res 2007: 21976.
-
Fosen KM, Thom SR (2014) Hyperbaric oxygen, vasculogenic stem cells, and wound healing. Antioxid Redox Signal 21: 1634-1647.
-
Veves A, Falanga V, Armstrong DG, Sabolinski ML (2001) Graftskin, a human skin equivalent, is effective in the management of noninfected neuropathic diabetic foot ulcers: a prospective randomized multicenter clinical trial. Diabetes Care 24: 290-295.
-
Villeneuve LM, Reddy MA, Natarajan R (2011) Epigenetics: deciphering its role in diabetes and its chronic complications. Clin Exp Pharmacol Physiol 38: 451-459.
-
Giacco F, Brownlee M (2010) Oxidative stress and diabetic complications. Circ Res 107: 1058-1070.
-
Berlanga-Acosta J, Mendoza-Mari Y, Martinez MD, Valdes-Perez C, Ojalvo AG, et al. (2013) Expression of cell proliferation cycle negative regulators in fibroblasts of an ischemic diabetic foot ulcer. A clinical case report. Int Wound J 10: 232-236.
-
Bouche C, Serdy S, Kahn CR, Goldfine AB (2004) The cellular fate of glucose and its relevance in type 2 diabetes. Endocr Rev 25: 807-830.
-
Roussel D, Dumas JF, Simard G, Malthi�ry Y, Ritz P (2004) Kinetics and control of oxidative phosphorylation in rat liver mitochondria after dexamethasone treatment. Biochem J 382: 491-499.
-
Taniguchi CM, Emanuelli B, Kahn CR (2006) Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7: 85-96.
-
Gemberling M, Bailey TJ, Hyde DR, Poss KD (2013) The zebrafish as a model for complex tissue regeneration. Trends Genet 29: 611-620.
-
Olsen AS, Sarras MP Jr, Leontovich A, Intine RV (2012) Heritable transmission of diabetic metabolic memory in zebrafish correlates with DNA hypomethylation and aberrant gene expression. Diabetes 61: 485-491.
-
Sarras MP Jr, Leontovich AA, Olsen AS, Intine RV (2013) Impaired tissue regeneration corresponds with altered expression of developmental genes that persists in the metabolic memory state of diabetic zebrafish. Wound Repair Regen 21: 320-328.
-
Sarras MP Jr, Leontovich AA, Olsen AS, Intine RV (2013) Impaired tissue regeneration corresponds with altered expression of developmental genes that persists in the metabolic memory state of diabetic zebrafish. Wound Repair Regen 21: 320-328.
