

International Journal of Diabetes and Clinical Research
Alpha- and Beta-Cell Proliferation and Extracellular Signaling - A Hypothesis
Cyril J Craven*
Queensland University of Technology, Brisbane, Australia
*Corresponding author:
Cyril J Craven, (Retired Lecturer, Queensland University of Technology, Brisbane, Australia, E-mail: cj.craven@qut.edu.au)
Int J Diabetes Clin Res, IJDCR-2-029, (Volume 2, Issue 2), Review Article; ISSN: 2377-3634
Received: February 12, 2015 | Accepted: March 29, 2015 | Published: April 01, 2015
Citation: Craven CJ (2015) Alpha- and Beta-Cell Proliferation and Extracellular Signaling � A Hypothesis. Int J Diabetes Clin Res 2:029. 10.23937/2377-3634/1410029
Copyright: © 2015 Craven CJ. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
A model is offered towards an understanding of the cellular communications of two major cell types of the pancreas. The beta and alpha cells of the pancreas are considered to be coupled by the insulin and glucagon hormones that they produce, respectively. In this model, insulin stimulates growth, especially cell division, of the alpha cells while glucagon similarly stimulates the beta cells. Of equal importance to the system is the concentration of the insulin:glucagon complex to which the beta and alpha cells are directly exposed in the pancreatic environment. Its measurement by the cells allows each coupled cell to gauge the activity of the other cell.
Keywords
Beta and alpha cells, Insulin, Glucagon, Insulin:glucagon couplet complex
Abbreviations
INS: Insulin, GLN: Glucagon, CC: Couplet Complex of INS and GLN, CTC: Couplet Trefones and Cells.
Introduction
The control of blood glucose occurs particularly by insulin and glucagon produced by secretion from the beta and alpha cells, upon stimulation by high or low levels of glucose, respectively. The emphasis of the studies of these hormones is usually on their effects on extrapancreatic cells, for example, insulin insensitivity in various body cells and the effect of glucagon on liver cells.
The intracellular events that potentially occur within beta-cells following receptor-mediated interaction by extracellular insulin, insulin-like growth factor-I and other stimulants leading to, in particular, the proliferation of rodent beta-cells, has been discussed [1]. The nutrient requirement with mTOR activation, along with the effects of epidermal and platelet-derived growth factors etc, has also been discussed [2]. Within these two recent articles and a third "Perspectives" article [3], glucagon is not considered a participant. The current article offers an alternate scenario involving glucagon and the alpha-cell with emphasis on paracrine activities of alpha and beta cells [4].
The hypothesis proposed is to assist in obtaining a better understanding of the pancreas and patient treatment. Our knowledge is not yet complete with glycemic instability a serious problem in patients [5] with only a vague understanding of the pathogenic pathway of Type 2 diabetes [6] and the enigma of the alpha-cells still remaining [7]. The model proposed here goes towards an understanding of the harmonisation of insulin and glucagon levels which occurs by the effect of these hormones on the adjacent cells of the pancreas.
A more generic model has already been proposed as a basis towards an understanding of multicellular organisation and cellular interactions within tissue cells [8]. The base of this model is the specific cellular communications formed between the two cell types initially produced by asymmetric cell division of precursor cells, followed by symmetric cell division of the two types of cells. These communications are reciprocal and one of the couplet molecules, produced by one cell type, stimulates the growth of the other cell type via a cell receptor. The couplet molecules form a complex and it is the levels of the individual molecules and the complex which control cell division. This generic model has been described with specific reference to various diseases including diabetes [8].
This model is here more specifically interpreted to the beta and alpha cells of the pancreas, with the hormones insulin and glucagon. In this specific model, the beta and alpha cells are examples of an "a-Cell" and an "i-Cell" of the generic model where these cells are produced by asymmetric cell division of precursor cells. The insulin and glucagon are examples of two interacting, cell-stimulating molecules produced by these cells that form a complex described in the generic model as a "Trefone Couplet Complex". A "Trefone" is a simple generic name used to describe cell-stimulating molecules such as insulin or glucagon, derived from couplet cells [8]. A pair of these molecules forms this couplet complex (TCC) which, together with the individual molecules, controls cell division.
The Model
The model described is illustrated in figure 1.
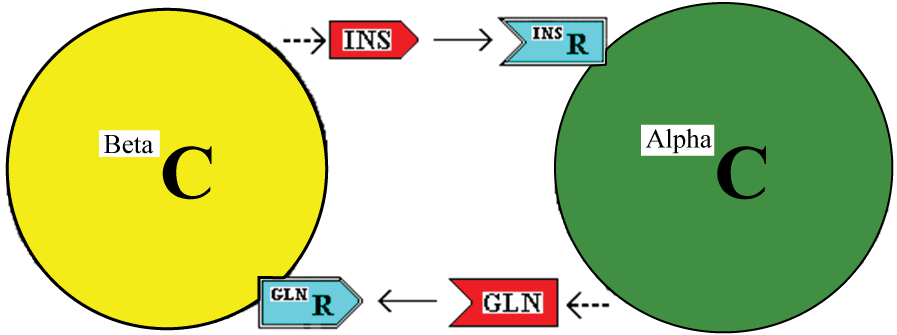
.
Figure 1: Interaction of Beta and Alpha cells (BetaC and AlphaC) through the stimulatory effects of insulin (INS) and glucagon (GLN) acting via cell membrane receptors.
View Figure 1
In this model, insulin and glucagon are stimulants of the alpha and beta cells, respectively. With adequate nutrient requirements available, they stimulate the recipient cells to grow in size and to produce more of the couplet stimulant. For example, insulin will stimulate the alpha cell to produce more glucagon. Further, each stimulant, sustained at a high level, along with adequate nutrients and other growth enhancers will bestow the competence to progress to cell division and to divide symmetrically or asymmetrically. This latter is contingent on the concentration of the insulin:glucagon complex. The calculated data relevant to this contingency is tabulated in Additional File Six of reference [8]. As an example, if the alpha cell detects a low level of free insulin, then the response depends on the concentration of the complex of insulin:glucagon. A low, medium or high concentration of complex would signal underactive, duly active or overactive status of the alpha cell itself which has produced low, medium and high levels of total glucagon respectively. The cell then has a measure of the amount of glucagon relative to the amount of insulin to allow a decision on whether the two hormones (and their source cells) are in harmony or not. If there is a sustained imbalance, a cellular decision, for each cell type, may then be to divide symmetrically, asymmetrically or with dedifferentiation or with transdifferentiation, as appropriate to bring the beta and alpha cells back into harmony. Apoptosis is also possible [8].
One possible means by which the cells could measure the insulin;glucagon complex would be to have cell membrane receptors for the complex. This is illustrated in figure 2. The existence of a membrane receptor is not integral to this hypothesis but there needs to be some intracellular mechanism to measure the level of complex [8].
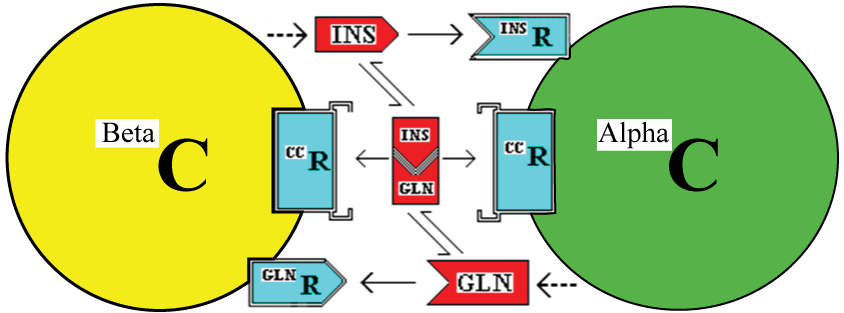
.
Figure 2: : Interaction of Beta and Alpha cells (BetaC and AlphaC) through the stimulatory effects of insulin (INS) and glucagon (GLN) acting via cell membrane receptors. In addition, this model incorporates a possible extracellular receptor for the Insulin:Glucagon complex (the CC in this case).
View Figure 2
Once internalised, a component or a signal derived from (i) the insulin, its receptor and/or the couplet complex in alpha cells or (ii) the glucagon, its receptor and/or the couplet complex in beta cells would need to localise to the nucleus to affect gene expression and cell division.
Main Evidence to Support the Model
For the proposed couplet cells (beta and alpha cells) with a couplet of insulin and glucagon, the following is expected:-
(1) Insulin binds glucagon
(2) (i) Beta cells have receptors for glucagon
(ii) Glucagon normally stimulates proliferation of beta cells
(iii) Glucagon inhibits proliferation of beta cells when both glucagon and insulin are high
(iv) Glucagon stimulates production/secretion of insulin by beta cells
(3) (i) Alpha cells have receptors for insulin
(ii) Insulin normally stimulates proliferation of alpha cells
(iii) Insulin inhibits proliferation of alpha cells when both insulin and glucagon are high
(iv) Insulin stimulates production/secretion of glucagon by alpha cells
(4) Each cell has a receptor for the insulin: glucagon complex.
The inhibitory effects are proposed to be associated with the high concentration of the insulin:glucagon complex. (See Additional File Six in reference [8].)
The evidence to support these expectations follow
(A) Insulin binds glucagon: This binding occurs but with apparent low affinity, with a Kd of 0.9 x 10-6, so it is said to have little significance physiologically importance [9]. However, this low binding could be enhanced in vivo by a metal ion. Zinc, which is known to bind insulin [10], is known to affect binary complex formation with a dramatic (8000-fold) increase in binding affinity [11], is known to be important in control of glucagon secretion and known to have a enhanced secretion (6.5 fold) from beta cells when exposed to glucose [7,12], could produce the required enhancement. In humans, the insulin is secreted as a hexamer with at least two atoms of zinc per hexamer [10] The formation of a ternary complex of zinc:insulin:glucagon could occur within the islets between cells as the blood flows from beta to alpha cells [13]. The interaction of glucagon and insulin with formation of the complex will also be more relevant because of the higher local concentrations of the hormones around the islets; the concentration may be 100 times that of plasma levels which are around 10-150 pmol/L. While this latter consideration is applicable in rodents and humans, it may not be in birds and horses where the alpha- and beta-cells are more separated
(B) (i) Beta cells have receptors for glucagon: Isolated rat pancreatic beta-cells have specific glucagon receptors as do a rat insulinoma cell line (INS-1) and a mouse insulinoma beta cell line (MIN6), while another tumour-derived beta-cell line (beta TC-3) does not express functional glucagon receptors [14-17].
(ii) Glucagon normally stimulates proliferation of beta cells: Overexpression of the glucagon receptor in pancreatic beta-cells increased both insulin content and α-cell mass (while not affecting islet cell organization or architecture) [18].
(iii) Glucagon inhibits proliferation of beta cells when [Glucagon] & [Insulin] are high: There may be no direct information on this effect when both are at "high" levels and when the concentration of the proposed complex (TCC) is also high and inhibitory. Within the current model, a high level of glucagon and of TCC would inhibit insulin production and secretion initially by a simple decrease in individual beta-cell release of insulin but, if the high levels were sustained, by a decrease in beta-cell proliferation.
(iv) Glucagon stimulates production/secretion of insulin by beta cell: Stimulation of insulin release by glucagon was observed in experiments on human subjects nearly 50 years ago - glucagon's insulinogenic effect [19,20]. Stimulation has also been shown to occur in isolated perfused rat pancreas [21], where concentrations of glucagon as low as 10-12 mol/L resulted in significant insulin release [15], and in cultured cloned pancreatic B cells (HIT-T15 and RINm5F) [22]. A high intracellular concentration of glucose is also a determinant of normal pancreatic insulin secretion.
(C) (i) Alpha cells have insulin receptors: The mRNA for the insulin receptor is expressed on clonal pancreatic alpha cells (glucagon-secreting cell lines eg. In-R1-G9 cells from an insulinoma, and alpha TC, clone 6 cells from an adenoma) and, in addition, labelled insulin is bound to receptors [23]. Electron microscopy has detected insulin on alpha cells in isolated rat islets [24].
(ii). Insulin stimulates proliferation of alpha cells: Insulin produced an up-regulation of α-cell proliferation in αTC1-6 cells (a mouse pancreatic α-cell line, a subclone of a glucagonoma-derived cell line) and this was via the insulin receptor [25]. Also, in diabetic mice with high insulin levels, both α-cell number and plasma glucagon levels increased as diabetes progressed [25].
(iii) Insulin inhibits proliferation of alpha cells when [Insulin] &[Glucagon] are high: There may be no direct information on this effect when both are at "high" levels and when the concentration of the proposed complex is also high and inhibitory. Within the current model, a high level of insulin and of TCC would inhibit glucagon production and secretion initially by a simple decrease in individual alpha-cell release of glucagon but, if the high levels were sustained, by a decrease in alpha-cell proliferation. Insulin is known to inhibit the proliferation of a certain melanoma cell line [26].
(iv) Insulin normally stimulates production/secretion of glucagon by alpha cells: At higher doses, perfused insulin has been shown to stimulate glucagon secretion in whole rats [27]. Also, the insulin receptor is required in alpha-cells for glucagon secretion induced by low glucose because siRNA-mediated "knockdown" of the insulin receptor in a pancreatic alpha-cell line (alpha-TC6), abolishes this glucagon secretion [28]. Evidence against the proposed model including that based on alpha cell-specific insulin receptor knockout mice [29], is discussed later.
The alpha and beta cells would each have a receptor for the insulin:glucagon complex
There is no information available on this. It is possible that one of the isoforms (IR-A or IR-B) of the insulin receptor could bind the insulin:glucagon complex (via the insulin component) or perhaps the insulin receptor-related receptor could do so, as it is known to be in pancreatic islets and likely in beta cells [30]. Beta cells also contain two glucagon receptors - one high- and one low-affinity for glucagon [31]. Perhaps the latter one could bind the glucagon:insulin complex better than it does free glucagon.
Further Evidence to Support (or Reject) the Model and General Discussion
Pulsatile secretions
The release of glucagon and insulin from human pancreatic islets is pulsatile. The kinetics of insulin and glucagon (and somatostatin) releases were studied in batches of 10-15 islets perfused with glucose. An increase of glucose from 3 to 20mmol/L resulted in a brief pulse of glucagon at one minute, followed by a pulse of insulin. Glucose continued to generate pulsatile releases of the hormones with 7- to 8-min periods but the repetitive glucagon pulses were anti-synchronous to pulses of insulin [32]. This could be interpreted in the current model as indicating that the first release of glucagon stimulates insulin, then insulin stimulates glucagon, and so on, where the respective receptors on the beta and alpha cells are momentarily exposed to hormones from the spiked transcellular fluid before this interstitial fluid is whisked away by perfusion fluid. In reality, plasma would contribute to the extracellular fluid and blood flow would rapidly remove the fluid containing the oscillating trefone concentrations. In this case, the cytoarchitecture of the perfused islets would need to allow some contact with alpha cells before contact with beta cells. Alpha cells are mainly on the periphery but scattered among the beta-cells in human islets [33].
Nuclear localization
The nuclear presence of insulin and glucagon (or their receptors) in alpha and beta cells respectively, is an expectation. Both insulin and its receptor have been identified within nuclei of hepatocytes [34] and insulin signals both to activate serine kinases within the nucleus directly and thus both to regulate a transcription factor and to stimulate the translocation of some active kinases to the nucleus in various cells [35]. However, there is no known study of the nuclear localization of insulin in alpha cells. Glucagon is internalized by hepatocytes [36] but its nuclear presence has not been reported and its localization in beta cells has not been studied.
Two receptors
The model explains the variability of the effects of insulin and glucagon by the presence of two receptors eg. one cell-membrane receptor for insulin and one for the insulin:glucagon complex. The binding site of the receptor for the complex could be very similar in structure to the receptor that binds insulin. Just as an antibody prepared against insulin could be expected also to bind to the insulin:glucagon complex (unless the epitope of insulin is obscured by the binding site of glucagon to the insulin), so there could be some receptor cross-reactivity between free insulin and the bound insulin. Receptor A may bind insulin with high affinity, while Receptor B, which binds insulin with low affinity, could bind the complex with high affinity, The evidence for two receptors, one with high affinity to bind free insulin and one with high affinity to bind the bound form, is not directly available but there is ample evidence for the existence of two receptors for many hormones and growth factors [8]. Often a receptor is said to have a high-affinity and low-affinity binding sites for a specific molecule but there may be two different receptors in the mix where the receptor with high affinity is the true receptor for the free molecule and the one with low affinity is in fact receptor for the complex.
Alpha cell-specific insulin receptor knockout
Knocking out the insulin receptor in mice alpha cells leads to increased glucagon secretion. This is not expected within the proposed model!
But there are unanswered questions within this study. In the "fed" state, glucose, insulin and glucagon are all significantly increased compared to control mice at two, five and 12 months of age (except for the insulin level at two months which is normal!). Pancreatic functions are obviously not normal - it is not purely a normal pancreas with normal alpha cells which happen to have no insulin receptors! Related questions pertain to this approach. How has the pancreas developed in the absence of the insulin receptor in alpha cells? One would expect that these cells would have been somewhat deprived of glucose (with no insulin receptors) throughout pancreatic development. Low levels of glucose would affect all metabolic reactions including production and secretion of glucagon by these alpha cells so all cells could be glucagon over-replete. One could assume that the normal presence of the insulin receptor was important and that changes in the embryonic development may have modified the alpha cells and other cells to compensate for the receptor's absence. Cell division and the embryonic development of the pancreas would have been affected via MTOR. Would the "alpha" cells in the islets then be "real" alpha cells? If the proposed model were correct, then the whole cellular infrastructure of the alpha cell would have been changed by removal of the insulin receptor. If the sole reason for the existence of an alpha cell is to produce glucagon, then it needs to monitor insulin levels and glucose levels - in the absence of an insulin receptor, the cells could not do this and would cease to be "alpha" cells.
Further, only 2% of the normal number of alpha cells is required for somewhat normal function [37]. Does one expect 100 % of alpha cells in a "knock-out" mouse to be without the receptor or could there be a residual of normal cells?
Other evidence supporting insulin as an inhibitor of glucagon secretion
The model offered here has insulin as a stimulator of glucagon secretion. However, others have provided data to support the concept that insulin is a direct inhibitor of glucagon secretion and a major positive contributor to this latter concept was the stimulation of glucagon secretion in the rat by guinea pig anti-porcine insulin serum [38]. The interpretation was that the absence of insulin allowed freed secretion of glucagon. But this increase in glucagon could be explained by other, perhaps indirect, factors [39]. With insulin deficiency, intracellular glucose levels and catabolism in alpha cells will be low and these will promote glucagon secretion (ii) the spread of antibody into other nearby cells will affect paracrine interactions, or (iii) there may be a receptor-mediated A cell response to anti-idiotypic antibodies. The latter antibodies may be present in antibody production against insulin [40]. Another possibility is an analytical error in glucagon immunoassay associated with immunoglobulins producing spurious results [41,42].
But other evidence from a cell line (In-R1-G9, a glucagon-producing alpha-cell line derived from a hamster glucagonoma) also indicates that insulin decreases steady-state glucagon [43,44]. Although insulin is widely considered to be a physiological suppressor of glucagon secretion, its mode of action is not clear [45]. It may be that this is an indirect effect of the subsequent change in glucose concentrations rather than a direct effect of insulin as it difficult to distinguish between the two effects. Or, part of insulin's ability to inhibit glucagon secretion may be via the ventromedial hypothalamus [46].
Some authors question the validity of the extrapolation of evidence from cell cultures. For example, beta-TC6 is a clonal beta islet cell line derived from insulinomas arising in transgenic mice expressing the SV40 T antigen gene [47]. Can evidence from this cell line be translated to the islet micro-organ which is complex both hormonally and in cellular organization [48]. "Although In-Rl-G9 cells can be considered as a useful model to study the molecular mechanisms of glucagon gene expression our results should be interpreted with reservations in regard to the transformed state of these cells" [43].
In the proposed model
In the normal pancreas, beta cells have insulin receptors [49] and insulin would be required by these cells to allow uptake of glucose. Normal glucose metabolism within the cells would be required for general metabolism but in particular for production and storage of proinsulin in secretory granules, ready for secretion of insulin. Insulin reception and its subsequent internalization by beta cells (not into granules) is part of this process which then allows the cell to assess whether glucose levels are too high and whether insulin should be secreted. Thus the internalized insulin would be a different pool from that synthetized by the cell and stored as proinsulin. In the current model, a high glucose level would only be permissive of insulin secretion; glucagon reception by the beta cell would be the determinant of the level of insulin secretion.
In the normal pancreas, alpha cells also have insulin receptors [23] and insulin would be required by these cells to allow uptake of glucose. Again, normal glucose metabolism within the cells would be required for general metabolism but here, in particular, for production and storage of glucagon in secretory granules, ready for secretion. Insulin reception by alpha cells is part of this process which then allows the cell to assess whether glucose levels are too low and whether glucagon should be secreted. In the current model, a low glucose level would only be permissive of glucagon secretion; insulin reception by the alpha cell would be the determinant of the level of glucagon secretion.
The relationships between glucose levels and the secretion of insulin and glucagon appears complex [50]. In mouse islets, at low glucose levels (0-7mmol/L), insulin secretion is low and constant while glucagon secretion is initially high at zero glucose (taken as 100% secretion) but reduces to ~30% at 7mmol/L. From 7-30mmol/L, insulin secretion increases as expected but glucagon levels unexpectedly also increase [50]. Each could be stimulating the secretion of the other in this isolated environment.
It should be noted that experiments that address insulin and glucagon interactions using exogenous additions of these hormones may be flawed if zinc is an essential component of the complex of insulin and glucagon. Certainly, zinc is secreted in excess of the two atoms of zinc which are associated with every hexamer of insulin [10].
General Discussion
In this model, insulin and glucagon are both considered to have dual activities in beta and alpha cells. They will have their normal metabolic actions on glucose metabolism as well as influencing cell-division decisions more directly, acting then as so-called trefones.
Insulin is required for glucose metabolic reasons in most/all cells and insulin receptors have been detected on beta cell lines RINm5F (an insulin-producing rat pancreatic cell line) [51] and B-TC3 (an insulin-secreting beta-cell, insulinoma line) [52]. While this evidence is from cell lines, it would be expected that as normal beta (and alpha) islet cells in the pancreas utilize glucose for energy purposes, they too would possess insulin receptors. However, within beta cells, insulin endocytosed by insulin receptors will not act as a trefone to affect cell-life decisions (outside of energetic considerations). Only in alpha cells will there be a trefone effect of insulin on proliferation via these insulin receptors - just as beta cells need glucagon receptors to control proliferation. Insulin receptor is more abundant in alpha-cells compared to beta-cells [53] as would be expected with a dual function in alpha cells.
In total then, glucagon will stimulate beta cells to produce insulin only if nutrients (glucose, fatty acids and/or certain amino acids) and consequently ATP in the cell are high. ATP may be responsible for so-called "nutrient signalling" [54,55]. Insulin will stimulate alpha cell to produce glucagon only if nutrients (glucose, fatty acids and/or certain amino acids) and consequently ATP in the cell are low. The effect of nutrients is controlled by MTOR (Mammalian target of rapamycin) which is a protein kinase that integrates signals from mitogens and nutrients to regulate cellular growth and proliferation. The insulin-stimulated α-cell proliferation acts through the mTOR signalling pathway [25].
The model offered here is simplified in that a variety of other hormones/growth factors and neurotransmitters are not considered but they do affect the release of insulin and glucagon by stimulation or inhibition of beta and alpha cellular activity [56]. Somatostatin, glucagon-like peptide and epinephrine are included in the list and these would need to be incorporated in more complex model that is needed for an understanding of the whole of the endocrine pancreas. Other molecules may also stimulate beta cell growth. For example, the incretin glucagon-like peptide-1 enhances beta-cell neogenesis, but is not here regarded as a direct stimulator of cell division of beta or alpha cells.
This simple model would require an asymmetric cell division in the embryonic development of the alpha and beta cells of the pancreas. A study of neogenesis of alpha and beta cells should indicate coexpression of glucagon and insulin, prior to asymmetric cell division(s) to produce the individual alpha and beta cells. After this cell division, the protein expression of each may depend on the immediate cellular environment such that gene activation may have occurred but without translation [57].
Information from microarray analysis [58] could possibly be zippered into the proposed model although the prevailing model of pancreatic development from the embryo has asymmetric cell division producing the alpha cell with a couplet beta/delta cell which subsequently produces the individual beta and delta cells. This would mean that these two cells would both produce insulin and they would be coupled by their own couplet trefones - perhaps somatostatin and amylin. As delta cells also produce glucagon, a complex interactive grouping of cells could evolve in the pancreas.
The model includes cellular measurement of the complex of insulin:glucagon where this determines whether the alpha and beta cells divide symmetrically, asymmetrically, with dedifferentiation or with transdifferentiation. The latter has been observed in conditions of depletion of beta cells, where alpha-cells in the islets transdifferentiate into beta-cells [59,60]. This is seen to be a new role of alpha-cells to replenish beta-cells; in the context of the current model, it would indicate the ability of alpha cells to detect an absence of beta cells as evaluated by low levels of insulin and insulin;glucagon complex. Transdifferentiation is on option included in Additional File Six of reference [8] when there is an imbalance of cells.
The importance of the insulin:glucagon ratio has been held by some researchers for over 40 years. It is considered that the ratio (if measured and acted upon by the body) would maintains glycemic stability even in extremes of glucose influx or efflux [61,62]. However, in contrast to the present model of insulin stimulating glucagon and vice versa, the proposal by the latter authors involves within-islet inhibition of alpha-cells by beta-cells ie. insulin suppresses glucagon secretion to create the required ratio [62]. To the contrary of this and not inconsistent with the present model, co-culture of an alpha cell line with a beta cell line produced increased amounts of both glucagon and insulin [25].
Some of the quoted evidence relies on data derived from cell line but it may be necessary to accept such evidence from cell lines with caution. "Any methodical intervention to measure changes may alter normal anatomy and physiology. For example, even if paracrine events were convincingly demonstrated in monolayer islet cell cultures, or dispersed islet cells, or isolated islets, the relevance to possible paracrine events in the complex islet micro-organ of the living intact cell is problematic" [48]. The lack of a valid model in diabetes research is reflected in the ongoing search for the perfect one. With limited success in 30 years, there is still an urgent need to establish a "normal" beta cell line [63] preferably of human origin.
Conclusion
The specific model proposed here is an example of the CTC model proposed on the basis of asymmetric cell division and division of labour [8]. Evidence is presented to support the basic tenet that insulin and glucagon are trefone couplets with the beta and alpha cells being the cell couplets. Glucagon stimulates both insulin secretion by, and proliferation of, beta cells via glucagon receptors. Reciprocally, insulin stimulates glucagon secretion by, and proliferation of, alpha cells via insulin receptors. These effects obviously occur within the organized architecture of the pancreatic lobes. It is suggested that, within the pancreas, where the local concentrations will be much higher than in plasma, and in the presence of zinc, the complex of insulin and glucagon will exist in a significant concentration. Reciprocating, pulsatile production of insulin and glucagon will occur within the pancreas reflecting the release from the couplet cells as they respond to the levels of their respective stimulating trefone and the insulin:glucagon complex.
The deficiency in insulin activity in diabetes may be due to insulin insensitivity in the presence of adequate insulin or due to insulin deficiency. Where there is a deficiency, this may be due to the rate of apoptosis surpassing the rate of proliferation from symmetric cell division of existing cells and from neogenesis from stem cells. In diabetes, the rate of apoptosis exceeds the rate of proliferation and neogenesis, all of which are higher than normal [64,65]. It is then the balance and control of apoptosis versus proliferation that is critical to maintaining normal cell mass and insulin production. It is then a balance between insulin and glucagon that controls metabolic imbalance leading to diabetes.
This basic model obviously does not take into account the somatostatin-secreting (D) cells, the pancreatic polypeptide-secreting (PP or F) cells nor the organized distribution of the cells among the four pancreatic lobes. Also, the effects of other interactive hormones etc from distant organs (epinephrine, glucocorticicoids, glucagon-like peptide) need to be incorporated into any overall understanding of pancreatic function.
References
-
Kulkarni RN, Mizrachi EB, Ocana AG, Stewart AF (2012) Human β-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes 61: 2205-2213.
-
Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, et al. (2014) Human β-cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes 63: 819-831.
-
Stewart AF, Hussain MA, Garc�a-Oca�a A, Vasavada RC, Bhushan A, et al. (2015) Human β-Cell Proliferation and Intracellular Signaling: Part 3. Diabetes 64: 1872-1885.
-
Hoang Do O, Thorn P (2015) Insulin secretion from beta cells within intact islets: location matters. Clin Exp Pharmacol Physiol 42: 406-414.
-
Bessho M, Murase-Mishiba Y, Imagawa A, Terasaki J, Hanafusa T (2014) Possible contribution of taurine to distorted glucagon secretion in intra-islet insulin deficiency: a metabolome analysis using a novel alpha-cell model of insulin-deficient diabetes. PLoS One 9: e113254.
-
Lee Y, Berglund ED, Yu X, Wang MY, Evans MR, et al. (2014) Hyperglycemia in rodent models of type 2 diabetes requires insulin-resistant alpha cells. Proc Natl Acad Sci U S A 111: 13217-13222.
-
Gromada J, Franklin I, Wollheim CB (2007) Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev 28: 84-116.
-
Craven CJ (2014) A model to explain specific cellular communications and cellular harmony:- a hypothesis of coupled cells and interactive coupling molecules. Theor Biol Med Model 11: 40.
-
Root-Bernstein RS, Dobbelstein C (2001) Insulin binds to glucagon forming a complex that is hyper-antigenic and inducing complementary antibodies having an idiotype-antiidiotype relationship. Autoimmunity 33: 153-169.
-
Lemaire K, Chimienti F, Schuit F (2012) Zinc transporters and their role in the pancreatic beta-cell. J Diabetes Investig 3: 202-211.
-
Cunningham BC, Bass S, Fuh G, Wells JA (1990) Zinc mediation of the binding of human growth hormone to the human prolactin receptor. Science 250: 1709-1712.
-
Ishihara H, Maechler P, Gjinovci A, Herrera PL, Wollheim CB (2003) Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat Cell Biol 5: 330-335.
-
Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE (1993) A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes 42: 1715-1720.
-
Moens K, Heimberg H, Flamez D, Huypens P, Quartier E, et al. (1996) Expression and functional activity of glucagon, glucagon-like peptide I, and glucose-dependent insulinotropic peptide receptors in rat pancreatic islet cells. Diabetes 45: 257-261.
-
Kieffer TJ, Heller RS, Unson CG, Weir GC, Habener JF (1996) Distribution of glucagon receptors on hormone-specific endocrine cells of rat pancreatic islets. Endocrinology 137: 5119-5125.
-
Yamato E, Ikegami H, Takekawa K, Fujisawa T, Nakagawa Y, et al. (1997) Tissue-specific and glucose-dependent expression of receptor genes for glucagon and glucagon-like peptide-1 (GLP-1). Horm Metab Res 29: 56-59.
-
Huypens P, Ling Z, Pipeleers D, Schuit F (2000) Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia 43: 1012-1019.
-
Gelling RW, Vuguin PM, Du XQ, Cui L, R�mer J, et al. (2009) Pancreatic beta-cell overexpression of the glucagon receptor gene results in enhanced beta-cell function and mass. Am J Physiol Endocrinol Metab 297: E695-707.
-
Samols E, Marri G, Marks V (1966) Interrelationship of glucagon, insulin and glucose. The insulinogenic effect of glucagon. Diabetes 15: 855-866.
-
Ahr�n B, Nobin A, Scherst�n B (1987) Insulin and C-peptide secretory responses to glucagon in man: studies on the dose-response relationships. Acta Med Scand 221: 185-190.
-
Kawai K, Yokota C, Ohashi S, Watanabe Y, Yamashita K (1995) Evidence that glucagon stimulates insulin secretion through its own receptor in rats. Diabetologia 38: 274-276.
-
Lambert DG, Atkins TW (1989) Effects of islet hormones on insulin secretion from cloned B cell lines, HIT-T15 and RINm5F. J Endocrinol 121: 479-485.
-
Kisanuki K, Kishikawa H, Araki E, Shirotani T, Uehara M, et al. (1995) Expression of insulin receptor on clonal pancreatic alpha cells and its possible role for insulin-stimulated negative regulation of glucagon secretion. Diabetologia 38: 422-429.
-
Patel YC, Amherdt M, Orci L (1982) Quantitative electron microscopic autoradiography of insulin, glucagon, and somatostatin binding sites on islets. Science 217: 1155-1156.
-
Liu Z, Kim W, Chen Z, Shin YK, Carlson OD, et al. (2011) Insulin and glucagon regulate pancreatic α-cell proliferation. PLoS One 6: e16096.
-
Kahn R, Murray M, Pawelek J (1980) Inhibition of proliferation of cloudman S91 melanoma cells by insulin and characterization of some insulin-resistant variants. J Cell Physiol 103: 109-119.
-
Dunbar JC, Walsh MF, Fo� PP (1978) The serum glucose response to glucagon suppression with somatostatin, insulin or antiglucagon serum in depancreatized rats. Diabetologia 14: 53-58.
-
Diao J, Asghar Z, Chan CB, Wheeler MB (2005) Glucose-regulated glucagon secretion requires insulin receptor expression in pancreatic alpha-cells. J Biol Chem 280: 33487-33496.
-
Kawamori D, Kurpad AJ, Hu J, Liew CW, Shih JL, et al. (2009) Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab 9: 350-361.
-
Ozaki K (1998) Insulin receptor-related receptor in rat islets of Langerhans. Eur J Endocrinol 139: 244-247.
-
Moens K, Flamez D, Van Schravendijk C, Ling Z, Pipeleers D, et al. (1998) Dual glucagon recognition by pancreatic beta-cells via glucagon and glucagon-like peptide 1 receptors. Diabetes 47: 66-72.
-
Hellman B, Salehi A, Gylfe E, Dansk H, Grapengiesser E (2009) Glucose generates coincident insulin and somatostatin pulses and antisynchronous glucagon pulses from human pancreatic islets. Endocrinology 150: 5334-5340.
-
Bonner-Weir S, O'Brien TD (2008) Islets in type 2 diabetes: in honor of Dr. Robert C. Turner. Diabetes 57: 2899-2904.
-
Podlecki DA, Smith RM, Kao M, Tsai P, Huecksteadt T, et al. (1987) Nuclear translocation of the insulin receptor. A possible mediator of insulin's long term effects. J Biol Chem 262: 3362-3368.
-
Kim SJ, Kahn CR (1997) Insulin regulation of mitogen-activated protein kinase kinase (MEK), mitogen-activated protein kinase and casein kinase in the cell nucleus: a possible role in the regulation of gene expression. Biochem J 323: 621-627.
-
Horwitz EM, Gurd RS (1988) Quantitative analysis of internalization of glucagon by isolated hepatocytes. Arch Biochem Biophys 267: 758-769.
-
Thorel F, Damond N, Chera S, Wiederkehr A, Thorens B, et al. (2011) Normal glucagon signaling and β-cell function after near-total α-cell ablation in adult mice. Diabetes 60: 2872-2882.
-
Maruyama H, Hisatomi A, Orci L, Grodsky GM, Unger RH (1984) Insulin within islets is a physiologic glucagon release inhibitor. J Clin Invest 74: 2296-2299
-
Samols E, Stagner JI, Ewart RB, Marks V (1988) The order of islet microvascular cellular perfusion is B----A----D in the perfused rat pancreas. J Clin Invest 82: 350-353.
-
Reilly TM, Root RT (1986) Production of idiotypic and anti-idiotypic antibodies by BALB/c mice in response to immunizations with glucagon, vasopressin, or insulin: supporting evidence for the network concept. J Immunol 137: 597-602.
-
Hattori N, Saiki Y, Kajikawa M, Ishihara T, Moridera K, et al. (1995) Immunoglobulin G can cross-react with glucagon antisera and cause a spuriously high plasma immunoreactive glucagon level. Endocr J 42: 455-460.
-
Yamamoto Y, Yamagishi S, Noto Y, Taniguchi M, Nagai Y, et al. (1996) The first case of insulin-dependent diabetes mellitus with prominent spurious hyperglucagonemia due to interference of immunoglobulin G in glucagon radioimmunoassay (OAL-123) system. Horm Res 45: 295-299.
-
Philippe J (1989) Glucagon gene transcription is negatively regulated by insulin in a hamster islet cell line. J Clin Invest 84: 672-677.
-
Kaneko K, Shirotani T, Araki E, Matsumoto K, Taguchi T, et al. (1999) Insulin inhibits glucagon secretion by the activation of PI3-kinase in In-R1-G9 cells. Diabetes Res Clin Pract 44: 83-92.
-
Bansal P, Wang Q (2008) Insulin as a physiological modulator of glucagon secretion. Am J Physiol Endocrinol Metab 295: E751-761.
-
Paranjape SA, Chan O, Zhu W, Horblitt AM, McNay EC, et al. (2010) Influence of insulin in the ventromedial hypothalamus on pancreatic glucagon secretion in vivo. Diabetes 59: 1521-1527.
-
Knaack D, Fiore DM, Surana M, Leiser M, Laurance M, et al. (1994) Clonal insulinoma cell line that stably maintains correct glucose responsiveness. Diabetes 43: 1413-1417.
-
Samols E, Bonner-Weir S, Weir GC (1986) Intra-islet insulin-glucagon-somatostatin relationships. Clin Endocrinol Metab 15: 33-58.
-
Bell GI, Polonsky KS (2001) Diabetes mellitus and genetically programmed defects in beta-cell function. Nature 414: 788-791.
-
Gylfe E, Gilon P (2014) Glucose regulation of glucagon secretion. Diabetes Res Clin Pract 103: 1-10.
-
Gazzano H, Halban P, Prentki M, Ballotti R, Brandenburg D, et al. (1985) Identification of functional insulin receptors on membranes from an insulin-producing cell line (RINm5F). Biochem J 226: 867-872.
-
Rothenberg PL, Willison LD, Simon J, Wolf BA (1995) Glucose-induced insulin receptor tyrosine phosphorylation in insulin-secreting beta-cells. Diabetes 44: 802-809.
-
Franklin I, Gromada J, Gjinovci A, Theander S, Wollheim CB (2005) Beta-cell secretory products activate alpha-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes 54: 1808-1815.
-
McDaniel ML, Marshall CA, Pappan KL, Kwon G (2002) Metabolic and autocrine regulation of the mammalian target of rapamycin by pancreatic beta-cells. Diabetes 51: 2877-2885.
-
Kwon G, Marshall CA, Pappan KL, Remedi MS, McDaniel ML (2004) Signaling elements involved in the metabolic regulation of mTOR by nutrients, incretins, and growth factors in islets. Diabetes 53 Suppl 3: S225-232.
-
Nielsen JH, Galsgaard ED, M�ldrup A, Friedrichsen BN, Billestrup N, et al. (2001) Regulation of beta-cell mass by hormones and growth factors. Diabetes 50 Suppl 1: S25-29.
-
Teitelman G, Alpert S, Polak JM, Martinez A, Hanahan D (1993) Precursor cells of mouse endocrine pancreas coexpress insulin, glucagon and the neuronal proteins tyrosine hydroxylase and neuropeptide Y, but not pancreatic polypeptide. Development 118: 1031-1039.
-
Chiang MK, Melton DA (2003) Single-cell transcript analysis of pancreas development. Dev Cell 4: 383-393.
-
Habener JF, Stanojevic V (2012) α-cell role in β-cell generation and regeneration. Islets 4: 188-198.
-
Paris M, Tourrel-Cuzin C, Plachot C, Ktorza A (2004) Review: pancreatic beta-cell neogenesis revisited. Exp Diabesity Res 5: 111-121.
-
Unger RH (1971) Glucagon and the insulin: glucagon ratio in diabetes and other catabolic illnesses. Diabetes 20: 834-838.
-
Unger RH, Orci L (2010) Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci U S A 107: 16009-16012.
-
Skelin M, Rupnik M, Cencic A (2010) Pancreatic beta cell lines and their applications in diabetes mellitus research. ALTEX 27: 105-113.
-
Rhodes CJ (2005) Type 2 diabetes-a matter of beta-cell life and death? Science 307: 380-384.
-
Ackermann AM, Gannon M (2007) Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. J Mol Endocrinol 38: 193-206.
