

International Journal of Diabetes and Clinical Research
Exogenous Phosphatidyl-Serine and -Ethanolamine Supports Anti-Proliferative and Chemoresistance Properties of ATAD3B and Induces S100B Recruitment at Mitochondria
Denis Rousseau*
Université Grenoble Alpes, Grenoble, France
*Corresponding author:
Denis Rousseau Inserm, Grenoble University, BP53 38041, Grenoble Cedex, France, Tel: (33) 04 76 63 56 00, Fax: (33) 04 76 51 42 18, E-mail: Denis.Rousseau@ujf-grenoble.fr
Int J Diabetes Clin Res, IJDCR-3-059, (Volume 3, Issue 2), Research Article; ISSN: 2377-3634
Received: January 07, 2016 | Accepted: May 24, 2016 | Published: May 27, 2016
Citation: Rousseau D (2016) Exogenous Phosphatidyl-Serine and -Ethanolamine Supports Anti-Proliferative and Chemoresistance Properties of ATAD3B and Induces S100B Recruitment at Mitochondria. Int J Diabetes Clin Res 3:059. 10.23937/2377-3634/1410059
Copyright: © 2016 Rousseau D. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
ATAD3 protein is a S100B- and PKC-regulated mitochondrial ATPase which is vital for mitochondrial biogenesis and early development. Even if suspected today to contribute in a lipid rafts transfer system, from endoplasmic reticulum to mitochondria, the precise function of ATAD3 is still unknown. However, ATAD3 has been previously shown to exhibits in vitro anti-proliferative and chemo resistant properties and to be a positive marker of different tumor subtypes, of tumor progression and of poor survival in several human cancers. This is particularly true concerning ATAD3B, the embryonic and cancer-expressed isoform. Because ATAD3 is hypothetically implicated in lipid fluxes between endoplasmic reticulum and mitochondria, like S100B itself, we asked whether ATAD3B anti-proliferative and chemo resistant effects are dependent on available phospholipids from extra-cellular source. Using ATAD3B expressing and control cells, we show here that ATAD3B-related biological functions are dependent on the lipids in the medium. Complementation of lipid-free medium with individual lipids shows that phosphatidyl-serine and phosphatidyl-ethanolamine specifically enhance the growth of ATAD3B over expressing cells.
Also, because S100B is a partner and regulator of ATAD3A/B, we investigated S100B localization following lipid starvation and exogenous phosphatidyl-serine delivery. We observed that endogenous S100B is first recruited to the plasma membrane and then to the mitochondria rapidly after phosphatidyl-serine addition. Finally, the in vivo mitochondrial localization of S100B was found in agreement with the in vitro interaction of S100B with mitochondria. These results reinforce the putative role of ATAD3 and S100B in Reticulum-Mitochondria interactions, like for the transport of phosphatidyl-serine between these compartments.
Keywords
ATAD3, S100B, Glioma, Chemoresistance, Doxorubicin, Phosphatidylserine
Introduction
ATAD3 is an inner membrane mitochondrial ATPase discovered in 2002, both as a c-Myc target gene [1], as an auto-immune tumor-related antigen and a marker of head and neck cancers [2,3]. ATAD3, especially ATAD3B (see below), was later shown to support chemoresistance in cancer cells and to induce cell cycle delay [4-9]. Also, and despite a still undiscovered function, ATAD3 is vital for mouse, drosophila and c. elegans early developments [10-14].
ATAD3 has been shown recently to promote MAM-mitochondria interactions and mitochondrial biogenesis fluxes, like the related transport of cholesterol and of phosphatidyl-serine [15-17]. In accordance with this, ATAD3 has been shown to be a limiting factor in mitochondrial biogenesis and adipogenesis [13-18].
ATAD3 exists as a single gene from pluri-cellular organisms until mammals, where the gene has duplicated twice in primates with 3 present genes (ATAD3A, 3B and 3C, where 3A corresponds to the ancestral gene). Among the three isoforms, ATAD3B has focused more attention because of its embryonic and cancer-associated expression profile [19]. This isoform has been early shown to be a very clear cancer marker [2,3,6,7,20,21], because ATAD3B is not expressed in adult healthy tissues. ATAD3B is today a prognostic marker for glial and breast cancers [9,22]. Up today, ATAD3B like ATAD3A has been shown over expressed in all studied cancers [8].
ATAD3A and ATAD3B have been shown to interact specifically with S100B and S100A1 [15,23]. S100B, like S100A1, is a calcium binding protein which participates in the regulation of various cellular functions, at different locations. S100B may act as a partner and regulator of PKC, then, ATAD3 could be among the various PKC substrates [6,24]. Also, no explanation can still justifies the various localizations of S100B, from the serum, the plasma membrane and the cytosol, to the mitochondria and the nucleus [25]. This can reflect the general role of S100B in PKC regulations. More recently, a new and interesting role of S100 protein has emerged and was proposed by Kuge and collaborators [26]. They showed that S100B can promote the transport of phosphatidyl-serine (PS) from the reticulum to the mitochondria for its further decarboxylation into phosphatidyl-ethanolamine (PE). Similar to the function of S100 at the plasma membrane lipid rafts, S100 could in theory regulate endoplasmic reticulum/mitochondria contact sites and lipid fluxes. In fact, it is well known that S100 plays a role in lipid rafting systems, like with the interplay between S100A10/Annexin and ANNAK at plasma membrane rafts [27-29].
All these accumulating observations led us to ask the question whether biological functions of ATAD3B, revealed by chemoresistance and induction of cell cycle delay, could be directly linked to phospholipid metabolism [18]. For this purpose, we used human oligodendroglial cell line expressing or not ATAD3B, the product of the embryonic and tumor-expressed human gene, to study the impact of ATAD3B on phospholipid metabolism.
For this purpose, we choose the strategy of lipid depletion (with lipid-free Fetal Bovine Serum) and of individual phospholipids delivery to test ATAD3B anti-proliferative and chemoresistance properties.
Materials and Methods
Cell culture
The human oligodendroglial-derived HS683 cell line [30], generously provided by G. Labourdette, was grown in DMEM supplemented with 10% fetal bovine serum (Biowest) at 37°C and 5% CO2. For lipid starvation, we used the same medium with 10% of lipid-free FBS (Sigma-Aldrich). Stably transfected cells were produced using 625 μg/ml of G418 (GIBCO-BRL) for selection and 200 μg/ml for growing reconstituted pools. The cell line was checked for the presence of 1pLOH [4].
Growth assays and chemoresistance assays
For growth rate measurements, cells were split at 104 cell/cm2 by triplicates and the number determined every day after rinsing twice with PBS and trypsin digestion. Cell suspensions were quantitated by automated counting with a calibration at 7 μm (coulter counter). For chemoresistance tests, cells were split at 105 cell/cm2 and treated continuously for 12 h with doxorubicin (Sigma) at the final concentration of 1.8, 5 or 25 μM. For counting of chemo resistant cells, cell cultures were rinsed twice in PBS to remove dying cells, treated with trypsin and re suspended in PBS. Cell suspensions were numerated by automated counting (coulter counter) with a calibration at 7 μm. Standard deviations were calculated with 16 measurements (four quadruplicates).
Mitochondria co-purification assay/western blot
Mitochondria were purified as described in [31]. There, after pelleting nuclei, the cytoplasm fraction is centrifuged at 100.000 g and wash three times the same way with extraction buffer. All fractions, cytoplasm, washes and mitochondria were analyzed by western-blot for the presence of co-purified S100B.
For western-blot, samples were lysed in SDS-sample buffer and protein concentration was measured with BCA™ protein assay kit (Pierce). Proteins were run on 12% SDS-PAGE gel, and transferred to a nitrocellulose membrane. Immuno blotting was performed in 0.2% Tween 20-TBS with the different antibodies (ATAD3 is a polyclonal antipeptide antibody from Eurogentec [4], S100B is a monoclonal antibody from Abcam (ab52642) and tubulin is a polyclonal antibody from Santacruz) and revealed with anti-mouse or anti-rabbit secondary antibodies coupled to HR peroxidase. Blots were revealed by chemo-luminescence according to the manufacturer's instructions (ECL, Amersham Biosciences).
Immunofluorescence confocal analysis
Detection of S100B was performed on HS683 cells overexpressing ATAD3B. Cells were seeded in the Lab-TekTM-Chamber slide system (Nunc, Danemark) and induced to differentiate for various time periods. Just before imaging, cells were incubated with 200 nM MitoTraker Green FM (Interchim, France) at 37°C in a 5% CO2 incubator for 30 min and 1 μg/ml Hoechst 33342 in the dark at 37°C for 10 min. After staining, cells were washed twice with pre-warmed phosphate-buffered saline (PBS), and fresh DMEM medium was added. Images were collected with a Leica TCS SP2 AOBS inverted laser scanning confocal microscope equipped with a 63X water immersion objective (HCX PL APO 63.0x/1,40 W Corr). Laser excitation was 351-364 nm for Hoechst, 488 nm for MitoTracker Green. Fluorescence emissions adjusted with AOBS were 390-470 nm for Hoechst and 498-549 nm for MitoTracker Green.
Statistics
Experiments were repeated at least three times and mostly analyzed in different parallels. Resulting data are given as means +/- standard deviation. Differences between data were analyzed by two-tailed, two-sample, unequal-variance Student’s t-test with P-levels indicated according to * for P < 0.05, ** for P < 0.01, *** for P < 0.001.
Results
ATAD3B-induced growth inhibition and chemoresistance depend on the lipids in the media
As described previously several times, ATAD3B over expression associates with cellular transformation but also induces growth inhibition and increased chemoresistance to genotoxics, like ATAD3A [4,9]. These phenomena have been well shown in glioma-derived cells but are especially visible in HS683 cell line which are oligodendroglioma-derived cells missing with both ATAD3B alleles [4].
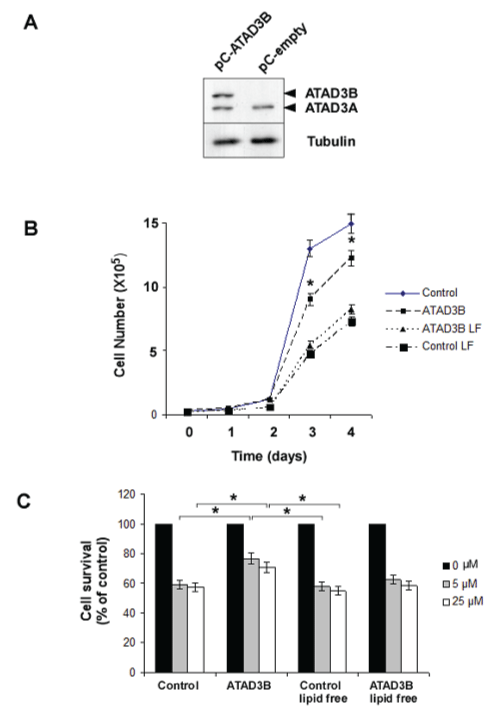
In order to see if ATAD3B expression is involved in lipid metabolism, we asked the inverse question whether lipid metabolism would influence ATAD3B properties and functions. To see if ATAD3B-induced growth inhibition is linked to lipid metabolism, like dependent of exogenous lipid source, we grew and compared the growth rate of ATAD3B expressing and control HS683 human cells under normal or lipid-free culture conditions (Figure 1A and Figure 1B). To avoid any clonal effects from selection, we used pools of transfected cells for both ATAD3B and control cells. As observed previously [4], ATAD3B expressing cells divided more slowly than control cells in normal media (division time of 29 h versus 20h). In lipid-free media (after 5 days of starvation), both the HS683 control cells and ATAD3B expressing cells grow slower but at nearly same speed (division time of 46 h). Since no more difference in growth speed were observed between both cells in these conditions, we concluded that the anti-proliferative properties of ATAD3B depend on lipids present in the medium.
.
Figure 1: (A) HS683 cells were stably transfected with ATAD3B-containing or empty pCDNA3 vectors. Western-blot detection of ATAD3 was performed to check for expression levels with tubulin as a control; (B) Growth of ATAD3B-expressing and control cells was measured during 4 days in presence or absence of lipids in the media (LF for lipid free medium); (C) Chemoresistance of ATAD3B-expressing and control cells to doxorubicin was measured in presence or absence of lipids in the media using two concentrations of doxorubicin (5 and 25 μM). Living cells were counted after 12 hours of treatment.
View Figure 1
In a similar way we looked if ATAD3B-induced chemoresistance to genotoxic is also dependent of exogenous lipid source (Figure 1C). As can be seen, ATAD3B expression increases short-term chemoresistance to doxorubicin in normal media as previously described [4], but no significant difference was observed under lipid starvation. Therefore, ATAD3B-induced chemoresistance, like cell cycle delay, are both dependent of lipid source in the media.
Exogenous lipids rescue cell growth in an ATAD3B-dependent manner
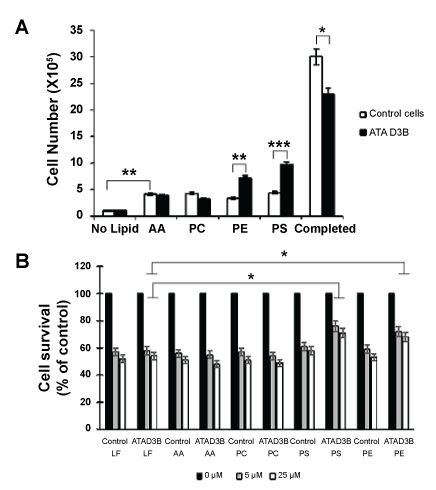
To further analyze this phenomenon, we used 5 days lipid-starved HS683 cells (ATAD3B-expressing and control) to add back individual lipids in the media (i.e., Arachidonic Acid, AA; Phosphatidyl-choline, PC; Phosphatidyl-ethanolamine, PE and Phosphatidyl-serine, PS) and to test if this affects the cell growth differentially (Figure 2A). As it can be seen, all lipids improved the cell growth of both cell lines, even if a slight cytotoxicity was observed soon after lipid addition (measured at 3% of cell death). Also, a significant difference was observed between control and ATAD3B expressing cells. Even if all lipids restored growth of control cells, PE and PS exhibit specifically a higher growth effect on ATAD3B expressing cells.
.
Figure 2: (A) Cell growth of ATAD3B-expressing and control cells was measured following restitution of individual lipids to 5 days lipid-starved cells. Three days after lipids addition, cell numbers were counted and are represented as a histogram as compared to no lipid or with complete medium; (B) Chemoresistance of ATAD3B-expressing and control cells to doxorubicin was measured on starved cells (LF) and on cells restituted with individual lipids. Living cells were counted after 12 hours of treatment and are represented as a histogram.
View Figure 2
We then concluded that ATAD3B expression improves PS- and PE-dependent cell growth and has no specific effect on AA- and PC-induced cell growth. These results confirm the link between ATAD3B expression level and exogenous PS/PE sources, naturally required for cell growth.
In a same way, we tested if adding back individual lipids to lipid-starved cells can restore ATAD3B-specific chemoresistance to doxorubicin (Figure 2B). As it could be seen, neither AA nor PC can modify chemoresistance to doxorubicin in both cell lines. Also, PS and PE addition lead to an increase of chemoresistance specifically in ATAD3B expressing cells. Therefore, it appears that ATAD3B-induced chemoresistance is functionally linked to PE and PS metabolism.
As introduced, ATAD3A/B has been first identified as targets of S100B, which is itself potentially involved in lipid metabolism. Therefore, we tried to see if endogenous S100B could be recruited at mitochondria under lipid starvation and/or PS-dependent cell growth.
S100B localizes at plasma membrane following lipid starvation and re-localizes to mitochondria after PS addition
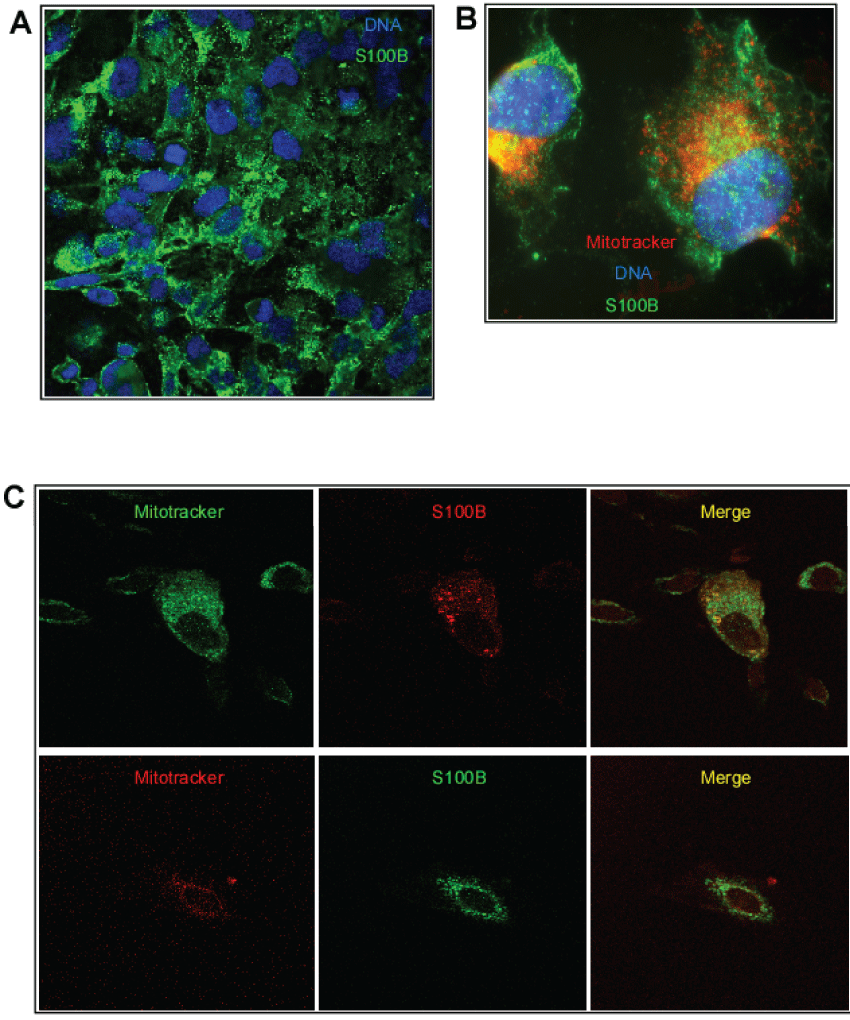
To look at S100B localization during lipid starvation and after PS addition, we performed confocal immuno-fluorescence microscopy analysis at different times using a specific anti-S100B monoclonal antibody (Figures 3). Under normal growing condition, in complete medium, S100B localizes everywhere in the cells (Figure 3A). Following lipid starvation, S100B localizes everywhere in the cells but some appear as intense dots at the cell surface, as presented as a whole cell view in figure 3B, but not co-localized with mitochondria. After 30 to 90 minutes of PS addition, an obvious accumulation of S100B was observed around mitochondria and demonstrated by confocal analysis of superimposed signal with mitotracker staining (Figure 3C).
.
Figure 3: (A) Stably transfected HS683 cells expressing ATAD3B were grown in normal media and imaged for S100B(whole cells view, DNA in blue-Hoechst) using green secondary antibody; (B) Stably transfected HS683 cells expressing ATAD3B were lipid-starved for 5 days and stained for mitochondria (Mitotracker-Red), DNA (Hoechst (blue) and S100B (green) and is presented as a whole cell view; (C) Stably transfected HS683 cells expressing ATAD3B were lipid-starved for 5 days and restituted with PS in the medium for 60 minutes. Fixated-permeabilized cells were stained with Mitotracker-Green (green or red) and immuno-stained for S100B (red or green) and imaged by confocal analysis.
View Figure 3
This result led us to the conclusion that S100B is recruited at the cell surface under lipid starvation and in part at mitochondria surface following PS-induced cell growth.
Phosphatidylserine addition induces S100B interaction with mitochondria
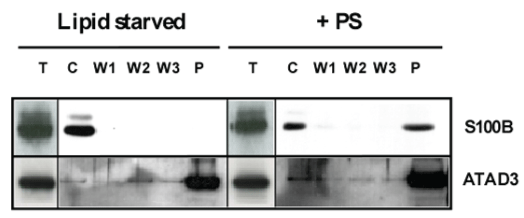
To confirm the re-localization of S100B at mitochondria after PS-induced cell growth, we performed the purification of mitochondria from lipid-starved and from PS-induced HS683 cells in order to analyze the presence of co-purified S100B (Figure 4). As it can be seen, S100B does not co-purify with mitochondria in control condition (lipid-starved) and is essentially present in the cytoplasmic sub-fraction. At the opposite, S100B does bind to mitochondria from PS-induced cells (almost half of the cytoplasmic S100B content). This binding is tight as no S100B was detected in the washing steps. ATAD3 immuno-detection (ATAD3A) was performed here to control the amount and integrity of mitochondria, as for S100B showing no change in expression. Also, similar results were obtained with ATAD3B-expressing cells (data not shown) but they cannot allow to discriminate the respective and direct roles of ATAD3A and ATAD3B. These observations confirm therefore that PS-induced cell growth is associated with the binding of S100B at mitochondria.
.
Figure 4: Mitochondria from lipid-starved and PS-induced (for 2 hours) HS683 cells were purified and washed 3 times with extraction buffer. Cytoplasm (C), washes (W) and pellets of mitochondria (P) were analyzed by Western-blot for the detection of S100B. ATAD3 was detected to check the efficiency of the procedure and the integrity of mitochondria. Total extracts were used as controls (T).
View Figure 4
Discussion
ATAD3 protein, even if vital for early development, has unknown molecular function up to date. However, three assays allow to reveal indirectly ATAD3A/B functions. One is the specific in vivo and in vitro binding to S100B [23], the second is the cell cycle delay induced by ATAD3A/B over expression [4] and the third is the associated chemoresistance to genotoxic which depends also on ATAD3A/B expression level [4-7,9].
We used here all the three methods to investigate the possible role of ATAD3B and S100B in lipid metabolism [15-17]. Speculating at start that ATAD3A/B may promote lipid-dependent cell growth, we observed that ATAD3B-induced anti-proliferative and chemoresistance properties are dependent of lipid source in the medium. Therefore, these characteristics of ATAD3A/B should be linked to the cellular lipid metabolism. We also found, by adding back individual phospholipids in the medium, that ATAD3B expression favors specifically the growth induced by PS and PE, showing also that ATAD3B enhances chemoresistance in presence of PS and PE.
Therefore, we focused on the localization of its partner S100B during the lipid starvation phase and after PS restitution. In normal cells, and as usually observed, S100B locates everywhere in the cells (as some to the mitochondria). After lipid starvation, we observed that S100B localizes partially to the plasma membrane and then to the mitochondria soon after PS delivery. The interaction of S100B to mitochondria was further confirmed by a mitochondria co-purification assay. Also, it is not sure that S100B binds to ATAD3A/B but it can be highly suspected [15,23]. However and all together, these results suggest that ATAD3B and S100B are involved in PS and PE intracellular metabolisms, like should be ATAD3A.
Considering S100B, we may believe that it can accompany PS and PE from the plasma membrane to the mitochondria, as a lipoprotein, or by controlling somewhere lipid transits as we know already the role of S100 in lipid rafts and lipid transfers [28]. Also, it is easy to believe that ATAD3, for its part, can contribute to the transfer of PS or PE from the reticulum compartment (ER) as it has been shown that ATAD3 is involved in the contact sites between ER and mitochondria [6] and in the transport of cholesterol through these points of contact [16]. Therefore, we think that ATAD3 can enhance lipid transport from ER to mitochondria and improve by this way the cell growth and chemoresistance. Of course, since ATAD3B interacts in a dominant-negative fashion with ATAD3A [15,32], we cannot exclude that both isoforms interact in vivo with S100B.
In conclusion, we speculate that increased circulating PS and PE may therefore improve ATAD3B chemoresistance properties, like could be for ATAD3A. Then, more practically and in the other way, decreased circulating levels of PS and PE might decrease ATAD3B-associated chemoresistance and may improve therapy of tumors like high-grade astrocytoma.
Acknowledgments
I greatly thank all the team of EMI104 laboratory (CEA-INSERM-Grenoble) for sharing facilities. I address a very special thanking to Yuliya Lyubenova Dimitrova for all her daily helps.
References
-
Zeller KI, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV (2003) An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol 4: 69.
-
Gires O, Münz M, Schaffrik M, Kieu C, Rauch J, et al. (2004) Profile identification of disease-associated humoral antigens using AMIDA, a novel proteomics-based technology. Cell Mol Life Sci 61: 1198-1207.
-
Schaffrik M, Mack B, Matthias C, Rauch J, Gires O (2006) Molecular characterization of the tumor-associated antigen AAA-TOB3. Cell Mol Life Sci 63: 2162-2174.
-
Hubstenberger A, Labourdette G, Baudier J, Rousseau D (2008) ATAD3A and ATAD3B are distal 1p-located genes differentially expressed in human glioma cell lines and present in vitro anti-oncogenic and chemoresistant properties. Exp Cell Res 314: 2870-2888.
-
Jiang Y, Liu X, Fang X, Wang X (2009) Proteomic analysis of mitochondria in Raji cells following exposure to radiation: implications for radiotherapy response. Protein Pept Lett 16: 1350-1359.
-
Fang HY, Chang CL, Hsu SH, Huang CY, Chiang SF, et al. (2010) ATPase family AAA domain-containing 3A is a novel anti-apoptotic factor in lung adenocarcinoma cells. J Cell Sci 123: 1171-1180.
-
Huang KH, Chow KC, Chang HW, Lin TY, Lee MC (2011) ATPase family AAA domain containing 3A is an anti-apoptotic factor and a secretion regulator of PSA in prostate cancer. Int J Mol Med 28: 9-15.
-
Li S, Rousseau D (2012) ATAD3, a vital membrane bound mitochondrial ATPase involved in tumor progression. J Bioenerg Biomembr 44: 189-197.
-
You WC, Chiou SH, Huang CY, Chiang SF, Yang CL, et al. (2013) Mitochondrial protein ATPase family, AAA domain containing 3A correlates with radioresistance in glioblastoma. Neuro Oncol 15: 1342-1352.
-
Piano F, Schetter AJ, Morton DG, Gunsalus KC, Reinke V, et al. (2002) Gene clustering based on RNAi phenotypes of ovary-enriched genes in C. elegans. Curr Biol 12: 1959-1964.
-
Kamath RS, Ahringer J (2003) Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30: 313-321.
-
Guertin DA, Guntur KV, Bell GW, Thoreen CC, Sabatini DM (2006) Functional genomics identifies TOR-regulated genes that control growth and division. Curr Biol 16: 958-970.
-
Hoffmann M, Bellance N, Rossignol R, Koopman WJ, Willems PH, et al. (2009) C. elegans ATAD-3 is essential for mitochondrial activity and development. PLoS One 4: e7644.
-
Goller T, Seibold UK, Kremmer E, Voos W, Kolanus W (2013) Atad3 function is essential for early post-implantation development in the mouse. PLoS One 8: e54799.
-
Gilquin B, Cannon BR, Hubstenberger A, Moulouel B, Falk E, et al. (2010) The calcium-dependent interaction between S100B and the mitochondrial AAA ATPase ATAD3A and the role of this complex in the cytoplasmic processing of ATAD3A. Mol Cell Biol 30: 2724-2736.
-
Rone MB, Midzak AS, Issop L, Rammouz G, Jagannathan S, et al. (2012) Identification of a dynamic mitochondrial protein complex driving cholesterol import, trafficking, and metabolism to steroid hormones. Mol Endocrinol 26: 1868-1882.
-
Issop L, Fan J, Lee S, Rone MB, Basu K, et al. (2015) Mitochondria-associated membrane formation in hormone-stimulated Leydig cell steroidogenesis: role of ATAD3. Endocrinology 156: 334-345.
-
Li S, Yao Y, Xu R, Pesenti S, Cottet-Rousselle C, et al. (2014) ATAD3 is a limiting factor in mitochondrial biogenesis and adipogenesis of white adipocyte-like 3T3-L1 cells. Mol Cell Biol.
-
Li S, Lamarche F, Charton R, Delphin C, Gires O, et al. (2014) Expression analysis of ATAD3 isoforms in rodent and human cell lines and tissues. Gene 535: 60-69.
-
Chen TC, Hung YC, Lin TY, Chang HW, Chiang IP, et al. (2011) Human papillomavirus infection and expression of ATPase family AAA domain containing 3A, a novel anti-autophagy factor, in uterine cervical cancer. Int J Mol Med 28: 689-696.
-
Merle N, Féraud O, Gilquin B, Hubstenberger A, Kieffer-Jacquinot S, et al. (2012) ATAD3B is a human embryonic stem cell specific mitochondrial protein, re-expressed in cancer cells, that functions as dominant negative for the ubiquitous ATAD3A. Mitochondrion 12: 441-448.
-
Ovaska K, Matarese F, Grote K, Charapitsa I, Cervera A, et al. (2013) Integrative analysis of deep sequencing data identifies estrogen receptor early response genes and links ATAD3B to poor survival in breast cancer. PLoS Comput Biol 9: e1003100.
-
Li S, Clémençon B, Catty P, Brandolin G, Schlattner U, et al. (2012) Yeast-based production and purification of HIS-tagged human ATAD3A, A specific target of S100B. Protein Expr Purif 83: 211-216.
-
Yáñez M, Gil-Longo J, Campos-Toimil M (2012) Calcium binding proteins. Adv Exp Med Biol 740: 461-482.
-
Deloulme JC, Raponi E, Gentil BJ, Bertacchi N, Marks A, et al. (2004) Nuclear expression of S100B in oligodendrocyte progenitor cells correlates with differentiation toward the oligodendroglial lineage and modulates oligodendrocytes maturation. Mol Cell Neurosci 4: 453-465.
-
Kuge O, Yamakawa Y, Nishijima M (2001) Enhancement of transport-dependent decarboxylation of phosphatidylserine by S100B protein in permeabilized Chinese hamster ovary cells. J Biol Chem 276: 23700-23706.
-
Gentil BJ, Delphin C, Mbele GO, Deloulme JC, Ferro M, et al. (2001) The giant protein AHNAK is a specific target for the calcium- and zinc-binding S100B protein: potential implications for Ca2+ homeostasis regulation by S100B. J Biol Chem 276: 23253-23261.
-
Benaud C, Gentil BJ, Assard N, Court M, Garin J, et al. (2004) AHNAK interaction with the annexin 2/S100A10 complex regulates cell membrane cytoarchitecture. J Cell Biol 164: 133-144.
-
Drücker P, Pejic M, Galla HJ, Gerke V (2013) Lipid segregation and membrane budding induced by the peripheral membrane binding protein annexin A2. J Biol Chem 288: 24764-24776.
-
Branle F, Lefranc F, Camby I, Jeuken J, Geurts-Moespot A, et al. (2002) Evaluation of the efficiency of chemotherapy in in vivo orthotopic models of human glioma cells with and without 1p19q deletions and in C6 rat orthotopic allografts serving for the evaluation of surgery combined with chemotherapy. Cancer 95: 641-655.
-
Hubstenberger A, Merle N, Charton R, Brandolin G, Rousseau D (2010) Topological analysis of ATAD3A insertion in purified human mitochondria. J Bioenerg Biomembr 42: 143-150.
-
Teng Y, Ren X, Li H, shull A, Kim J, et al. (2016) Mitochondrial ATAD3A combines with GRP78 to regulate the WASF3 metastasis-promoting protein. Oncogene 35: 333-343.
