

International Journal of Diabetes and Clinical Research
Activity-Induced Deamidation of Triose-Phosphate Isomerase May Explain the Deleterious Effects of Excessive Glucose Consumption
Alan R Hipkiss*
Aston Research Centre for Healthy Ageing (ARCHA), School of Health and Life Sciences, Aston University, UK
*Corresponding author:
Alan R Hipkiss, Aston Research Centre for Healthy Ageing (ARCHA), School of Health and Life Sciences, Aston University, Birmingham B4 7ET, United Kingdom, E-mail: alanandjill@lineone.net
Int J Diabetes Clin Res, IJDCR-3-066, (Volume 3, Issue 3), Review Article; ISSN: 2377-3634
Received: September 02, 2016 | Accepted: November 23, 2016 | Published: November 25, 2016
Citation: Hipkiss AR (2016) Activity-Induced Deamidation of Triose-Phosphate Isomerase May Explain the Deleterious Effects of Excessive Glucose Consumption. Int J Diabetes Clin Res 3:066. 10.23937/2377-3634/1410066
Copyright: © 2016 Hipkiss AR. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
It is suggested that the glycolytic enzyme triose-phosphate isomerase (TPI) is the "Achilles' heel" in carbohydrate metabolism under conditions of excessive glycolysis. The catalytic activity of TPI can induce permanent changes in its structure and a decline in activity. Under conditions of high glycolytic flux, insufficient TPI activity may eventually cause the accumulation of dihydroxyacetone phosphate (TPI substrate) which spontaneously decomposes into methylglyoxal (MG), a highly reactive bicarbonyl whose actions provoke many of the deleterious macromolecular changes associated with ageing. In cells in which synthesis of new TPI molecules is impossible (e.g. erythrocytes), and under conditions of excessive and persistent glycolysis, MG formation would be particularly enhanced, which could be released into the circulation.
Triose-Phosphate Isomerase, an Unusual Catalyst
According to the Oxford English Dictionary a catalyst is defined as a "substance that does not itself change, but speeds up a reaction". Enzymes, being biological catalysts, are similarly assumed to remain unaltered as a result of their catalytic action, and for the vast majority this definition appears valid. There is, however, at least one exception, the enzyme triose-phosphate isomerase (TPI). TPI is a member of the well characterized pathway of glucose catabolism (glycolysis) in which the 6-carbon sugar is converted into two 3-carbon molecules (pyruvic acid) and the energy released is collected in the form of the universal energy compound, adenosine triphosphate (ATP). TPI catalyses the conversion of the triose-phosphate dihydroxyacetone phosphate (DHAP) into glyceraldehyde-3-phosphate [G3P] or the reverse during gluconeogenesis. It is important to note that (i) not only are both these triose-phosphates reactive glycating agents, capable of reacting with proteins, mostly via available amino and guanidine groups of lysine and arginine residues respectively, but (ii) they can both spontaneously decompose into the highly reactive bicarbonyl, methylglyoxal (MG), which is increasingly regarded as responsible for much macromolecular modification, including cross-linking, which characterises the aged phenotype, as well as the secondary complications of persistent hyperglycaemia and type-2-diabetes [1-3].
More than two decades ago it was found that TPI, whilst performing its catalytic action, tends to undergo a chemical change. Gracy and co-workers were the first to observe that two asparagine residues (15 and 71) in TPI showed increasing potential for spontaneous deamidation following catalytic function [4-7]. In fact, it was shown that asparagine deamidation was enhanced by repeated catalytic activity, and, as a result, the dimeric TPI dissociated into monomers which decreased catalytic activity due to their enhanced susceptibility to proteolytic destruction by intracellular proteases. Indeed it has been concluded that "the probability of deamidation of an individual TPI molecule is a function of the number of times it is used as a catalyst" [8].
Asparagine residues in proteins are especially susceptible to spontaneous deamidation, generating any one of four different products; L-aspartic, L-iso-aspartic, D-aspartic and D-iso-aspartic acid residues. These changes are known to accompany aging of long-lived proteins such as lenticular crystallins, collagen and histones (see papers by Truscott for authoritative account) [9,10]. Although deamidation can also be induced when proteins are heated, there have been no reports of activity-induced enzyme deamidation other than that of TPI, with the possible exception of glucosephosphate isomerase [8].
As noted above, the consequences of asparagine residue deamidation will be a change in the primary structure of the proteins, possibly involving the presence of a D-amino acid and/or the addition of an extra carbon atom into the polypeptide backbone should an iso-aspartic acid residue be generated. As a result, the enzyme called protein iso-aspartate methyl transferase (PIMT) has evolved which facilitates conversion of the iso-form back to the meso-form ensuing a partial repair of the modified protein. Indeed PIMT deficiency is associated with profound neurological dysfunction and accelerated aging [11-14]. It is important to note that PIMT does not catalyse the re-amidation of the newly generated aspartyl residue.
Triose-phosphate Isomerase and a High Carbohydrate Diet
Many of the procedures which delay cellular and organism aging (dietary restriction, every-other-day feeding, treatment with non-metabolizable forms of glucose, rapamycin treatment, insulin-like growth hormone defects) involve decreased glycolytic function, which suggests that the glycolytic pathway is not entirely benign. In fact, much age-related dysfunction seems to be strongly associated with enhanced glucose catabolism resulting in polypeptide glycation and formation of advanced glycated end-products (AGEs), which are thought to be responsible for the secondary modifications which accompany hyperglycaemia and type-2 diabetes, most probably due to excess MG formation [13-15]. It may also be relevant to note that much age-related deterioration and disease occurs as a consequence of mitochondrial dysfunction. It is certainly possible that under circumstances of inadequate mitochondria-mediated ATP generation, upregulation of glycolytic activity occurs in compensation. This may help to explain why genetically determined mitochondrial defects in conditions such as Parkinson's disease result in enhanced protein glycation [15-17].
Consequently, it is proposed that TPI may be a metabolic "Achilles' heel" which, if over-used as a consequence of continuous glycolysis due to either mitochondrial dysfunction or incessant carbohydrate consumption, results in formation of MG as a result of the enzyme's failure to convert DHAP sufficiently rapidly into G3P and thereby permitting DHAP accumulation and its decomposition into MG. This would be particularly important in those cells and tissues in which synthesis of new TPI molecules is impossible, such as erythrocytes and cells of the eye lens core. The fact that TPI activity in erythrocytes appears to greatly exceed that of any other glycolytic enzyme [18] may represent an evolutionary adaptation to activity-induced decline in TPI catalytic function. That human erythrocytes have a fixed lifespan of around 120 days may also be a consequence of activity-induced TPI dysfunction.
That the diet of "modern" man frequently contains much higher levels of carbohydrate compared to the lower level to which we presumably evolved to consume, argues that, evolutionary, humans are not well adapted enzymatically to a high carbohydrate diet. Thus the current Western diet, rich in glucose-generating polysaccharide, especially if of a high glycaemic index (i.e. rapidly releasing glucose after ingestion), coupled with activity-induced decline in TPI function, may be the cause of MG-mediated molecular modification (e.g. AGEs) and MG-induced dysfunction such as in type-2 diabetes, neurodegeneration, atherosclerosis, cataractogenesis, nephrology and ageing generally. Figure 1 is a diagrammatic illustration of (i) the pivotal role of TPI activity in suppressing DHAP accumulation, and (ii) how loss of TPI, especially in cells unable to synthesise replacement molecules (e.g. erythrocytes and lens core cells) will eventually provoke MG accumulation and protein glycation if glyoxalase activity is insufficient due to age-related decline.
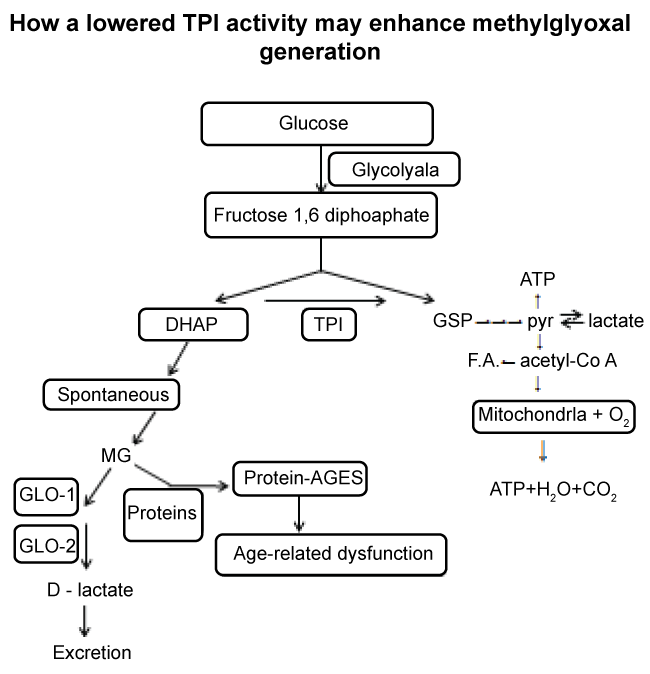
.
Figure 1: Illustration of the pivotal role of TPI in normally preventing DHAP accumulation thereby suppressing MG and protein-AGE formation.
Lowered activity of TPI in cells incapable of synthesising replacement molecules will allow DHAP accumulation and its decomposition into MG which, in absence of sufficient glyoxalase activity, will react with proteins to form protein -AGEs.
TPI = Triose-Phosphate Isomerase, GLO-1 = Glyoxalase-1 Activity, GLO-2 = Glyoxalase-2 Activity, MG = Methylglyoxal, DHAP = dihydroxyacetone phosphate, G3P = glyceraldehyde-3-phosphate.
View Figure 1
Conclusion
It is suggested that TPI may be the"Achilles'heel" in carbohydrate metabolism: excessive glycolysis, occurring as a result of high carbohydrate (and high glycaemic index) intake, can result in a decline in TPI activity and the increased generation of MG in quantities which over-whelm the detoxifying activity of glyoxalases-1 and -2 (See [19] for detailed discussion of these enzymes). Furthermore, because human erythrocytes cannot synthesize replacement TPI molecules, it is possible to suggest that erythrocytes could be a major source of circulatory MG, especially in conditions of persistent hyperglycaemia, with consequential deleterious effects throughout the body.
Conflict of Interest
The author declares no conflict of interest.
Acknowledgement
The author thanks Wendy Overton for help with the diagram.
References
-
Srikanth V, Westcott B, Forbes J, Phan TG, Beare R, et al. (2013) Methylglyoxal, cognitive function and cerebral atrophy in older people. J Gerontol A Biol Sci Med Sci 68: 68-73.
-
Allaman I, Bélanger M, Magistretti PJ (2015) Methylglyoxal, the dark side of glycolysis. Neurosci 9: 23.
-
Rabbani N, Xue M, Thornalley PJ (2016 a) Methylglyoxal-induced dicarbonyl stress in aging and disease: first steps towards glyoxalase 1-based treatments. Clin Sci (Lond) 30: 1677-1696.
-
Gracy RW, Talent JM, Zvaigzne AI (1998) Molecular wear and tear leads to terminal marking and the unstable isoforms of aging. J Exp Zool 282: 18-27.
-
Sun AQ, Yüksel KU, Gracy RW (1992) Relationship between the catalytic center and the primary degradation site of triosephosphate isomerase: effects of active site modification and deamidation. Arch Biochem Biophys 293: 382-390.
-
Ugur I, Aviyente V, Monard G (2012) Initiation of the reaction of deamidation in triosephosphate isomerase: investigations by means of molecular dynamics simulations. J Phys Chem B 116: 6288-6301.
-
De la Mora-de la Mora I, Torres-Larios A, Enríquez-Flores S, Méndez ST, Castillo-Villanueva A, et al. (2015) Structural effects of protein aging: terminal marking by deamidation in human triosephosphate isomerase. PLoS One 10: e0123379.
-
Robinson N E, Robinson AB (2004) Molecular Clocks: deamidation of asparaginyl and glutaminyl residues in peptides and proteins. Althouse Press, Oregon, USA, 231.
-
Truscott RJ, Schey KL, Friedrich MG (2016) Old Proteins in Man: A Field in its Infancy. Trends Biochem Sci 41: 654-664.
-
Truscott RJ (2010) Are ancient proteins responsible for the age-related decline in health and fitness? Rejuvenation Res 13: 83-89.
-
Qin Z, Dimitrijevic A, Aswad DW (2015) Accelerated protein damage in brains of PIMT+/- mice; a possible model for the variability of cognitive decline in human aging. Neurobiol Aging 36: 1029-1036.
-
D'Angelo S, Trojsi F, Salvatore A, Daniele L, Raimo M, et al. (2015) Accumulation of altered aspartyl residues in erythrocyte membrane proteins from patients with sporadic amyotrophic lateral sclerosis. Neurochem Int 63: 626-634.
-
Desrosiers RR, Fanelus I (2011) Damaged proteins bearing L-isoaspartyl residues and aging: a dynamic equilibrium between generation of isomerized forms and repair by PIMT. Curr Aging Sci 4: 8-18.
-
Bidinosti M, Martineau Y, Frank F, Sonenberg N (2010) Repair of isoaspartate formation modulates the interaction of deamidated 4E-BP2 with mTORC1 in brain. J Biol Chem 285: 19402-19408.
-
Salahuddin P, Rabbani G, Khan RH (2014) The role of advanced glycation end products in various types of neurodegenerative disease: a therapeutic approach. Cell Mol Biol Lett 19: 407-437.
-
Plotegher N, Bubacco L (2016) Lysines, Achilles' heel in alpha-synuclein conversion to a deadly neuronal endotoxin. Ageing Res Rev 26: 62-71.
-
Vicente Miranda H, El-Agnaf OM, Outeiro TF (2016) Glycation in Parkinson'sdisease and Alzheimer's disease. Mov Disord 31: 782-790.
-
Newsholme EA, Start C (1973) Regulation in Metabolism. John Wiley & Sons (London): 98-99.
-
Rabbani N, Xue M, Thornalley PJ (2016) Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj J 33: 513-525.
