

International Journal of Neurology and Neurotherapy
D-Leucine Suppresses Prion Formation in Prion-Infected Culture Cells
Kana Miyashita, Morika Suzuki, Kana Nishijima and Naomi Hachiya*
Department of Neurophysiology, Tokyo Medical University, Japan
*Corresponding author:
Naomi Hachiya, Department of Neurophysiology, Tokyo Medical University, 6-1-1 Shinjuku, Tokyo 1608402, Japan, Tel: +81-3-3351-6141; E-mail: naomi@tokyo-med.ac.jp
Int J Neurol Neurother, IJNN-1-011, (Volume 1, Issue 1), Short Communication; ISSN: 2378-3001
Received: September 20, 2014 | Accepted: November 20, 2014 | Published: November 24, 2014
Citation: Miyashita K, Suzuki M, Nishijima K, Hachiya N (2014) D-Leucine Suppresses Prion Formation in Prion-Infected Culture Cells. Int J Neurol Neurother 1:011. 10.23937/2378-3001/1/1/1011
Copyright: © 2014 Miyashita K, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Prion disease is an infectious and fatal disease. The pathogen consists of an abnormal form of the prion protein; designated PrPSc. PrPSc is insoluble, highly resistant to digestion by proteases and all disinfectants. In contrast, the cellular form of prion protein PrPC is easily soluble and digested by proteases. Direct interaction between PrPSc and PrPC is believed to induce the propagation of prions; however, the molecular mechanisms underlying this step have not been clarified. Despite efforts to find molecules that can specifically interact with the prion protein conformer and thereby inhibit prion production through disruption of PrPSc and PrPC interaction, and thus may disturb prion production by inhibiting the above interaction, there is no efficient way to date and data regarding this issue are limited. Our findings provide novel insight into possible therapeutic approaches for treating prion disease.
Keywords
Prion, Prion protein, D-amino acid, Propagation, Prion disease
Introduction
Prion diseases are lethal neurodegenerative disorders involving the misfolding of the host encoded cellular prion protein, PrPc to PrPSc [1]. PrPc is detergent soluble and proteinase K (PK)-sensitive, whereas PrPSc is detergent insoluble and partially PK- resistant [2]. These differences in biophysical properties are thought to be due to different conformations of the two isoforms. The former molecule is highly a-helical, while PrPSc contains a large proportion of ß-sheet structures [3,4]. Although PrPSc is thought to a pathogenic agent of disease, the molecular mechanism underlying PrPc to PrPSc conversion remains elusive.
Genetic types of the disease in humans comprise familial CJD (fCJD), fatal familial insomnia (FFI), prion protein cerebral angiopathy (PrP CAA) and Gerstmann-Straussler-Scheinker syndrome (GSS), while sporadic types include sporadic CJD (sCJD), sporadic fatal insomnia (sFFI) and variably protease-sensitive prionopathy (VPSPr) [5], the most recently identified prionopathy [6]. A rapidly expanding body of evidence also indicates that prions cause many different neurodegenerative disorders, including Alzheimer's and Parkinson's diseases as well as the frontotemporal dementias and some forms of amyotrophic lateral sclerosis (ALS) [7,8]. Several molecules such as quinacrine [9], antibodies [10] or pentosan sulfate [11] have demonstrated the ability to eliminate PrPSc, however, these agents have shown no or modest treatment efficacy in clinical trials [12]. Meanwhile, recent evidence has shown that significant concentrations of D-amino acids exist naturally even in the central nervous system and have demonstrated important physiological functions [13-15]. Furthermore, they have been exhibited to strongly prevent biofilm formation by bacteria and can also destroy existing biofilms due to their ability to breakdown amyloid fiber [16,17].
Based on this background, we have examined whether D-amino acids are capable of destroying PrPSc amyloid using a cell culture system.
Materials and Methods
Cell culture: Mouse neuroblastoma 2a (N2a) cells and the cells infected with the mouse adapted prion strain 22Lwere maintained at 37°C in 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM)(Sigma-Aldrich) [18].
D-amino acid treatment and assay for the PrPSc-degrading efficacy: D- and L- amino acids were purchased from Wako Pure Chemical Industries, Ltd. D-amino acid-dependent PrPSc-degrading efficacy was measured by western blot analysis. Following incubation for a limited period of time with the specified concentration of D- or L-amino acids, the cell lysates were incubated with proteinase K (1mg/ml) for one hour at 37°C, at which time digestion was stopped with the addition of PMSF at a final concentration of 1mM. Finally, 1% (w/v) of SDS was added and the proteins were methanol precipitated at -80°C for at least two hours. After centrifugation at 1,800xg for 30 minutes, the protein pellets were subjected to SDS-PAGE and western blotting using a PVDF membrane (Millipore). The membranes were incubated with the monoclonal primary antibody SAF83 (SPI Bio) followed by horseradish peroxidase-conjugated anti-mouse IgG as the secondary antibody (GE Healthcare). The membranes were subsequently developed with ECL-prime (GE Healthcare) and analyzed using a Versa Doc 5000MP (Bio-Rad).
Results and Discussion
In this study, the D-amino acid anti-prion amyloid-breaking activity was examined using an assay to determine the PrPSc levels in ScN2a cells infected with scrapie strain 22L. As indicated in Figure 1a, without PK treatment, PrP was detected as a broad band (Figure 1a, panel b, lane 1). In contrast, in the cells treated with PK, PrPSc was detected as three bands exhibiting the modification of zero, one or two sugar residues, respectively (Figure 1a, panel a, lane 1). Meanwhile, N2a cells that were not infected by prions did not show resistance to PK and clearly disappeared after the application of proteases (Figure 1a, panel a, lane 2).
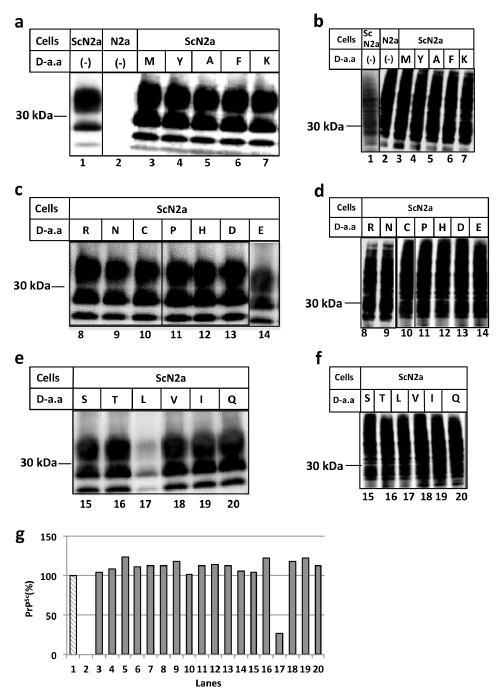
Figure 1(A): D-leucine has prion amyloid breaking activity
(Panels a, c, e): PrPSc levels in N2a (lane 2) or ScN2a (lanes 1, 3-7) cells were measured by PK treatment and western blotting after 24 hours incubation in the
presence of D-amino acids (1.0mM). The amino acids added in each lane are as follows: lane 3: Met, lane 4: Tyr, lane 5: Ala, lane 6: Phe, lane 7: Lys, lane8: Arg,
lane 9: Asn, lane 10: Cys, lane 11: Pro, lane 12: His, lane 13: Asp, lane 14: Glu, lane 15: Ser, lane 16: Thr, lane 17: Leu, lane 18: Val, lane 19: Ile, lane 20: Gln;
(Panels b, d, f): N2a or ScN2a cells were cultured for 24 hours in the presence of D-leucine at various concentrations and detected by western blotting without PK
treatment. (Panel g): Densitometric measurement of PrPSc bands of panels a, c and e
View Figure 1(A)
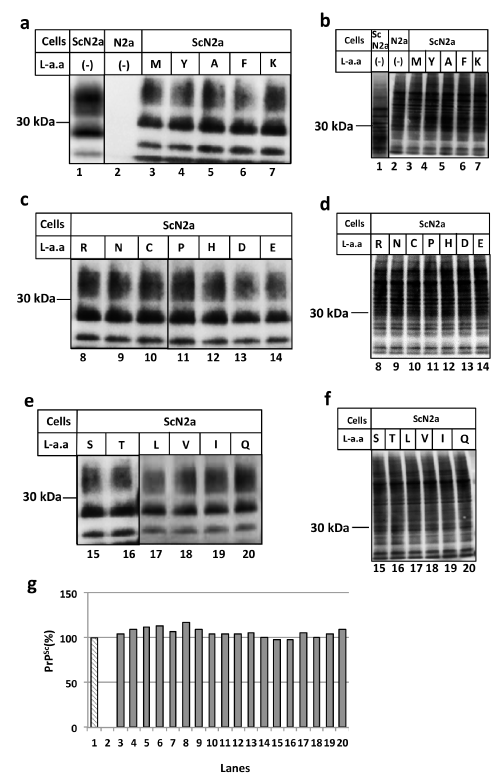
Figure 1 (B): :L-amino acid has no effect on prion conversion.
Each L-amino acid was added to ScN2a cells under the same experimental conditions as A.
View Figure 1(B)
1M of D-amino acids was added to ScN2a cell cultures directly and the cells were incubated at 37°C for 24 hours. After the incubation, cells were treated with (Figure 1a, panel a, lanes 3-7, panels c and e) or without (Figure 1a, panel b, lanes 3-7, panels d and f) proteinase K (PK). As shown in Figure 1a, D-leucine specifically reduced the amount of PrPSc, indicating the suppression of prion formation (Figure 1a, panel e, lane 17, panel g, lane 17) and this suppressive activity was increased in a dose-dependent manner (Figure 2, pane la). We also examined the amyloid-breakdown activity of L-amino acids under the same conditions as Figure 1a, however, there was no discernible effects (Figure 1b, Figure 2, panel b). Incidentally, D-tryptophan exhibited cytotoxicity and resulted in cell death after incubation, therefore further experiments using D-tryptophan were omitted (data not shown). The anti-prion activity of D-leucine peaked at 48 hours of incubation, subsequently becoming inactivated at 72 hours, (Figure 3, panel a) suggesting that D-leucine was metabolized. This effect was not seen with L-leucine incubation (Figure 3, panel b).
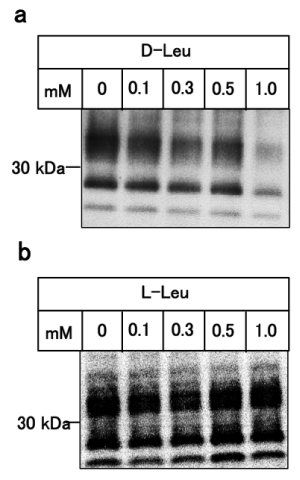
Figure 2: Dose-dependency of the anti-prion activity of D-leucine.
ScN2a cells were treated for 24 hours with various concentrations of
D-leucine (panel a) or L-leucine (panel b). Levels of PrPSc were analyzed
after PK digestion of cellular lysates by immunoblotting.
View Figure 2
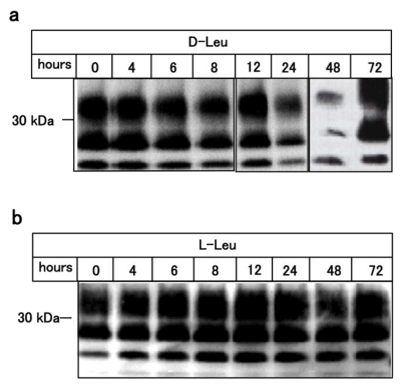
Figure 3: D-leucine reduces levels of protease resistant PrPScin a timedependent manner.
ScN2a cells were treated with D-leucine (panel a) or L-leucine (panel b) with
various incubation times. Levels of PrPSc were analyzed after PK digestion of
cellular lysates by immunoblotting.
View Figure 3
Despite an intensive search for therapeutic compounds for prion disease, none have significantly increased the survival of patients. Their effectiveness is often limited by their restricted access across the blood-brain-barrier (BBB), and no potential candidate has been identified to date that is effective after an infection has been established in the central nervous system [19,20].
Notably, all amino acids, except for glycine, are able to alter their conformation into two isometric L- and D-forms, as they have the potential to form two stereoisomers around a central carbon atom. D-amino acids were once thought to be an unusual state that exists only in the cell walls of bacteria. However, the development of experimental procedures has revealed that D-amino acids are present widely in organisms, even in mammals, including humans [13]. Based on these points of view, we have examined if D-amino acids possess the ability to breakdown the PrP-amyloid by using prion-infected ScN2a cells as a measure of proteinase K resistance.
Consequently, the present findings demonstrated that D-leucine has a strong specific anti-prion activity, which subsequently increases the sensitivity of ScN2a cells to proteinase K, resulting in the clearance of the pathogenic form of prion protein PrPSc. The inhibitory effect on prion formation by D -leucine shown in this study is most readily explained by the binding of D-leucine to PrPSc molecules on the cell surface, leading to interference of PrPc to PrPSc interaction. This binding may also be interfering with the aggregation process itself. Although some D-amino acids can exert an isomer-toxic effect [21,22], they exist naturally in dietary proteins and in foods [23], indicating that they are probably harmless. It remains to be established how decreased resistance to proteolysis results in reduced levels of PrPSc in ScN2a cells by D-leucine and the in vivo anti-prion activity of D-leucine remains to be determined using mice inoculated with infectious prion. Based on our finding, D-leucine, together with previous findings, would be an interesting prominent candidate for the therapeutic approach of prion disease.
Acknowledgement
We thank Y. Komata, C. Uchida and H. Kato for technical assistance. This work was supported by Grants-in-Aid from the Research Committee of Prion Disease, the Ministry of Health, Labor and Welfare of Japan.
References
-
Prusiner SB (1991) Molecular biology of prion diseases. Science 252: 1515-1522.
-
Meyer RK, McKinley MP, Bowman KA, Braunfeld MB, Barry RA, et al. (1986) Separation and properties of cellular and scrapie prion proteins. Proc Natl Acad Sci U S A 83: 2310-2314.
-
Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, et al. (1991) Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 30: 7672-7680.
-
Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, et al. (1993) Conversion of alpha helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 90: 10962-10966.
-
Aguzzi A, Calella AM (2009) Prions: Protein aggregation and infectious diseases. Physiol Rev 89: 1105-1152.
-
Zou WQ, Puoti G, Xiao X, Yuan J, Qing L, et al. (2010) Variably protease-sensitive prionopathy: A new sporadic disease of the prion protein. Ann Neurol 68: 162-172.
-
Jucker M, Walker LC (2011) Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol 70: 532-540.
-
Prusiner SB (2012) Cell biology. A unifying role for prions in neurodegenerative diseases. Science 336: 1511-1513.
-
Meyer zu HG, Mausberg AK, Korth C, Stuve O, Kieseier BC (2010) Quinpramine--a promising compound for treating immune-mediated demyelination of the nervous system. Drug News Perspect 23: 287-294.
-
Lauren J (2014) Cellular prion protein as a therapeutic target in Alzheimer's disease. J Alzheimers Dis 38: 227-244.
-
Rainov NG1, Tsuboi Y, Krolak-Salmon P, Vighetto A, Doh-Ura K (2007) Experimental treatments for human transmissible spongiform encephalopathies: is there a role for pentosan polysulfate?. Expert Opin Biol Ther 7: 713-726.
-
Sim VL, Caughey B (2009) Recent advances in prion chemotherapeutics. Infect Disord Drug Targets 9: 81-91.
-
Fujii N (2005) D-amino acid in elderly tissues. Biol Pharm Bull 28: 1585-1589.
-
Sasabe J, Suzuki M, Imanishi N, Aiso S (2014) Activity of D-amino acid oxidase is widespread in the human central nervous system. Front Synaptic Neurosci 10: 6-14.
-
Wolosker H, Dumin E, Balan L, Foltyn VN (2008) D-amino acids in the brain: D-serine in neurotransmission and neurodegeneration. FEBS J 275: 3514-3526.
-
Kolodkin-Gal I, Romero D, Cao S, Clardy J, Kolter R, et al. (2010) D-amino acids trigger biofilm disassembly. Science. 30: 627-629.
-
Leiman SA, May JM, Lebar MD, Kahne D, Kolter R (2013) D-amino acids indirectly inhibit biofilm formation in Bacillus subtilis by interfering with protein synthesis. J Bacteriol 195: 5391-5395.
-
Kishida H, Sakasegawa Y, Watanabe K, Yamakawa Y, Nishijima M, et al. (2004) Non-glycosylphosphatidylinositol (GPI)-anchored recombinant prion protein with dominant-negative mutation inhibits PrPSc replication in vitro. Amyloid 11: 14-20.
-
Nunziante M, Gilch S, Schatzl HM (2003) Prion diseases: from molecular biology to intervention strategies. ChemBioChem 4: 1268- 1284.
-
Weissmann C, Aguzzi A (2005) Approaches to therapy of prion diseases. Annu. ReV Med 56: 321-344.
-
Friedman M, Gumbmann MR, Masters PM (1984) Protein-alkali reactions: chemistry, toxicology, and nutritional consequences. Adv Exp Med Biol 177: 367-412.
-
Soto A, Del Raso NJ, Schlager JJ, Chan VT (2008) D-Serine exposure resulted in gene expression changes indicative of activation of fibrogenic pathways and down-regulation of energy metabolism and oxidative stress response. Toxicology 14: 177-192.
-
Friedman M, Levin CE (2012) Nutritional and medicinal aspects of D-amino acids. Amino Acids 42: 1553-1582.
