

International Journal of Pathology and Clinical Research
Anemia Investigation Reveals a Primary Sea-Blue Histiocyte Syndrome
António Proença Caetano1*, Inês de Figueiredo1, Francisco Tortosa2, Anabela Ferrão3 and Cristina Ferreira2
1Centro Hospitalar Lisboa Central and Faculty of Medicine, University of Lisbon, Portugal
2Assistant Professor at Institute of Pathology, Faculty of Medicine, University of Lisbon, and Hospital Santa Maria, Centro Hospitalar Lisboa Norte, Portugal
3Assistant Professor at Department of Pediatrics, Faculty of Medicine, University of Lisbon, and Hospital Santa Maria, Centro Hospitalar Lisboa Norte, Portugal
*Corresponding author:
António Proença Caetano, Centro Hospitalar Lisboa Central and Faculty of Medicine, University of Lisbon, Rua da Beneficiencia n°8, 1069-166 Lisboa , Portugal, Tel: 00351 913 746 170, E-mail: aprocaetano@gmail.com
Int J Pathol Clin Res, IJPCR-2-019, (Volume 2, Issue 1), Case Report and Review of Literature; ISSN: 2469-5807
Received: October 27, 2015 | Accepted: January 04, 2016 | Published: January 08, 2016
Citation: Caetano AP, de Figueiredo I, Tortosa F, Ferrão A, Ferreira C (2016) Anemia Investigation Reveals a Primary Sea-Blue Histiocyte Syndrome. Int J Pathol Clin Res 2:019. 10.23937/2469-5807/1510019
Copyright: © 2016 Caetano AP, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Sea-blue Histiocyte Syndrome (SBHS) is a rare and poorly understood systemic histiocytosis that is sometimes associated with haematological and lipid storage diseases as well as other miscellaneous conditions, but in most cases its cause is unknown. Patients often have very disparate clinical features but share the same histological findings of sea-blue histiocytosis in the bone marrow, i.e. characteristic lipid-laden macrophages with deep-blue or blue-green granules when stained with Romanovsky or May-Grunwald Giemsa stain. We describe a case of an 8 year-old girl admitted to the emergency department with a history of fever, adynamia, subicteric sclerotics, lymphadenopathies, anemia and leukopenia. During the investigation, histological examination of the bone marrow revealed a Sea blue histiocytosis. Patient workup revealed a spherocytosis, but it was inconclusive regarding major causes for these findings such as Niemann-Pick Disease, Gaucher Disease and other haematologic conditions, and was thus considered a Primary Sea-Blue Histiocyte Syndrome. A review of the literature was then carried out to outline current knowledge of this condition and to establish a rationale for the patient workup and management when such findings are encountered.
Keyword
Sea-Blue Histiocyte Syndrome, Anemia investigation
Introduction
Sea-blue Histiocyte Syndrome (SBHS) is a rare systemic histiocytosis first described in 1970 by Silverstein et al. [1], characterized by splenomegaly in association with the presence of numerous histiocytes in the bone marrow containing cytoplasmic granules that stain a deep azure blue with May-Grunwald Giemsa stain [1-3]. SBHS is generally associated with lipid storage diseases and/or haematological conditions, but the pathogenesis of the syndrome as well as the contents of the stored material observed in the histiocytes are still not well established [4].
Sea-blue histiocytes are commonly found in the bone marrow of patients with SBHS, but they may also occur in the spleen2, liver, lymph nodes and tonsils [5]. The etiology of the sea-blue histiocytes is unknown, although several authors link its formation to abnormal lipid metabolism since it is frequently accompanied by lipid storage disorders or diseases with excessive ineffective turnover of haematopoietic cells [6-8]. The clinical course is generally benign, although there have been descriptions of cases of disseminated involvement and extensive infiltration of vital organs such as the heart, lungs and liver with poor prognosis and outcome [9].
Sea-blue histiocytes were first identified by Sawitsky in 1947, where he described a 29 year-old man with unexplained splenomegaly and subsequently identified the presence of macrophages with closely packed granules in the spleen coloured deep blue azure with May-Grunwald stain. Also in 1947, Moeschlin [2] observed the presence of splenic macrophages that stained a deep azure blue with May-Grunwald stain, which he termed "sea-blue histiocytes". Similar cellular morphology was found in the bone marrow by Wewalka [3] in 1950. Further in 1954, Sawitsky described two young adults with sea-blue histiocytes present in the spleen and bone marrow and suggested that these combined findings might constitute a syndrome [10], but it was only in 1970 that Silverstein et al. used the term "sea-blue histiocyte syndrome" to refer to this entity [1].
Clinical Summary
We present a case of an otherwise healthy 8 year-old girl with no previous history of infection, asthenia, anorexia or loss of weight, with several previously reported episodes of pneumonia during childhood, two of them requiring hospitalization (the latest in 2010), dietary diversity successfully implemented, no known food or drug allergies or intolerance and vaccine plan up to date. She was the product of a normal pregnancy with 41 weeks gestation, eutocic delivery, Apgar 9 and 10 for 1st and 5th minute, respectively, and presented with hyperbilirubinemia (maximum value of 21.4 mg/dl (reference values < 1.0 mg/dl)) on the 2nd day of life, successfully treated with phototherapy (regular bilirubin levels achieved on day 4). Growth patterns were unremarkable with average weight between 75th and 90th percentiles, stature between 25th and 50th percentiles and cephalic perimeter remained between 75th and 90th percentiles. Motor, cognitive, social and language development were also unremarkable.
The patient was admitted to Hospital Torres Vedras, in Torres Vedras showing symptoms of prolonged fever (maximum of 39°C, which partially responded to antipyretics), food vomiting and frontal headaches for the past 3 weeks. Physical examination revealed subicteric sclerotics. She was treated symptomatically for acute gastroenteritis and discharged, but due to persistence of symptoms she returned to the hospital, where laboratory examination revealed Haemoglobin (Hb) levels of 5.4 g/dL (reference values 13.0-17.5 g/dL), white blood cell (WBC) count 3.9 × 109/L (reference values 4.0-11.0 × 109/L) with 78.8% neutrophils (reference value 45-54%) and 9.6% lymphocytes (reference value 16-25%), platelet count 128 × 109/L (reference value 150-450 × 109/L), normal coagulation and renal function tests, direct and indirect bilirrubin count of 3.79 mg/dL (reference value < 0.50 mg/dL) and 0.45 mg/dL respectively, lactate dehydrogenase of 583 IU/L (reference value 208-378 IU/L) and C Reactive Protein (CRP) 2.01 mg/dL (reference value < 0.50 mg/dL).
Physical examination
The patient was transferred to Hospital Santa Maria, in Lisbon, where physical examination revealed generalized weakness, cutaneous pallor, hydrated but pale mucosas, subicteric sclerotics, small bilateral nontender cervical lymphadenopathies, cardiac auscultation with a systolic murmur II/VI, and pulmonary auscultation with no remarkable changes. Abdominal examination detected a palpable spleen 3 cm below the abdominal rib margin and bilateral, elastic, fluctuant and nontender inguinal lymphadenopaties. She was apirectic and eupneic and her heart rate and blood pressure were within normal values. The remaining examination showed no relevant findings.
Lab data on admission
Repeated lab work at Hospital Santa Maria revealed haemoglobin 5.3 g/dl (reference value 13.0-17.5 g/dL), haematocrit 13.3% (reference value 40.0-50.0%), mean corpuscular volume (MCV) 70.9 fL (refrence value 80.0-97.0 fL), mean corpuscular haemaglobin (MCH) 28.4 pg (reference value 27.0-33.0 pg), WBC count 3.34 x109/L (reference value 4.0-11.0 x109/L) with 74.5% neutrophils (reference value 45-54%) and 17.0% lymphocytes (reference value 16-25%), platelet count 160 × 109/L (reference value 150-450 x 109/L), total bilirrubin levels 3.79 mg/dL (reference value < 1.0 mg/dL), aspartate transaminase (AST) 56 IU/L (reference value 0-34 IU/L), alanine transaminase (ALT) 23 IU/L (reference value 10-49 IU/L), LDH 1133 IU/L (reference value 208-378 IU/L), CRP 1.8 mg/dl (reference value < 0.50 mg/dL), blood glucose levels 95 mg/dL (reference value 70-110 mg/dL), urea 50 mg/dL (reference value 10-50 mg/dL), creatinine 0.5 mg/dL (reference value < 0.70 mg/dL), Na+ 133 mEq/L (reference value 135-145 mEq/L), K+ 3.6 mEq/L (reference value 3.5-5.1 mEq/L), urine osmolality 279 mOsml/Kg (reference value 275-295 mOsml/Kg), uric acid 4.1 mg/dL (reference value 3.7-9.2 mg/dL), total cholesterol 111 mg/dL (reference value < 190 mg/dL), high-density lipoprotein (HDL) 36 mg/dL (reference value > 40 mg/dL), low-density lipoprotein (LDL) 55 mg/dL (reference value < 110 mg/dL), triglycerides 102 mg/dL (reference value < 150 mg/dL).
Imagiology and additional tests
An abdominal ultrasound was performed and revealed hepatomegaly with regular contours and homogenous parenchyma, gallbladder stones (maximum size of 12 mm), without biliary tract ectasia, and homogeneous enlargement of the spleen (14.5 cm longitudinal axis). Pancreas and kidneys showed no remarkable changes and there was no evidence of ascitic fluid or enlarged lymph nodes.
Peripheral blood smear identified spherocytes with absence of blast cells. Direct Coombs test was negative; osmotic fragility studies were positive and glucose stress test did not reveal relevant changes. Haemoglobin electrophoresis revealed an HbA2 of 2.7% and HbF of 4.4%.
Bone marrow aspirate was performed and revealed a myeloid/erythroid shift in favor of the granulocytic series. Furthermore, megakaryocyte number was increased (1% of total cellularity), lymphocytes represented 22% and a hyper basophilic cytoplasm was observed in 2% of total cellularity. There was no evidence for morphologic changes or maturation shifts in the granulocytic and erythroid series. No atypical cells were identified.
Bone marrow biopsy reported a diffuse infiltration by histiocytic cells with vast cytoplasm and fine granules that stained blue with Giemsa stain and were PAS positive; they were negative for Ziehl-Neelsen stain (Figure 1 and Figure 2). This infiltration was accompanied by slight, moderately focal increase of the reticulin network. The hematopoietic elements present were unremarkable. The overall findings were compatible with Sea-Blue Histiocyte Syndrome, possibly associated with other diseases.
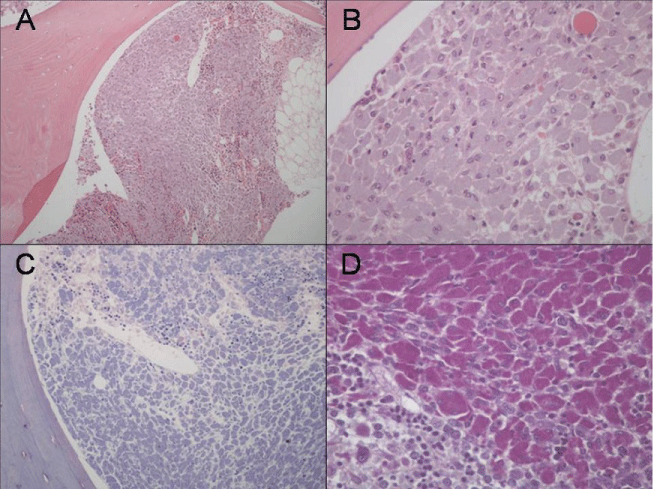
.
Figure 1: "Sea-Blue" granules found in macrophages of bone marrow biopsy. (A) Bone Marrow infiltration with histiocytic cells with vast cytoplasm (original magnification ×4). Hematoxylin and eosin stain; (B) The cells contain "sea-blue" granules (original magnification ×20). The granules are positive for Giemsa; (C) (original magnification ×4), Periodic-Acid Schiff; (D) (original magnification ×20), Panoptic and Grocott (not shown). The granules are negative for Ziehl-Neelsen and Perls stain (not shown). Optical Microscope Nikon Eclipse 50i. Images were captured with coupled digital camera DS Camera Control unit DS-L2. Institute of Pathology, Faculty of Medicine, University of Lisbon, and Hospital Santa Maria, Centro Hospitalar Lisboa Norte. The pathology exam was performed by two different pathologists with experience in bone marrow: franciscotortosa.pathology@gmail.com.
View Figure 1
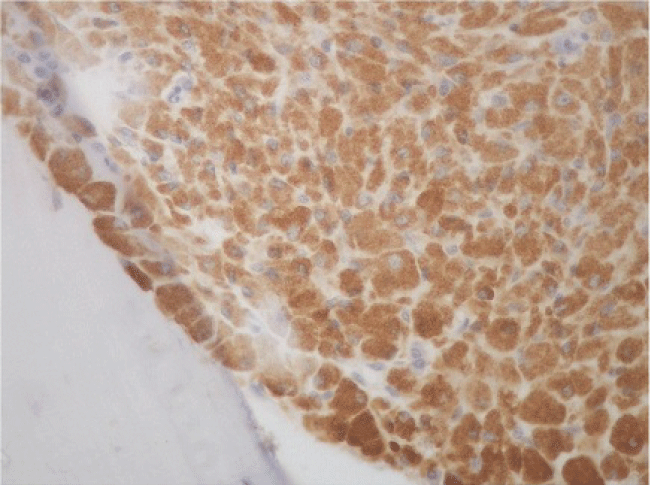
.
Figure 2: Immunohistochemical study with CD68 PG (original magnification ×20) demonstrates the histiocytic nature of the cells- Optical Microscope Nikon Eclipse 50i. Images were captured with coupled digital camera DS Camera Control unit DS-L2. Institute of Pathology, Faculty of Medicine, University of Lisbon, and Hospital Santa Maria, Centro Hospitalar Lisboa Norte. The pathology exam was performed by two different pathologists with experience in bone marrow: franciscotortosa.pathology@gmail.com.
View Figure 2
Lymphocyte populations were analyzed but showed no significant changes (CD3+ 86%, CD4+ 49,1%, CD8+ 32%). Quantitative immunoglobulin levels showed increase in IgM levels. Measurement of Glucose-6-Phosphate Dehydrogenase levels was normal (11.9 units/g).
Serology for Ebstein-Barr Virus (IgG positive), Cytomegalovirus, Adenovirus, Mycoplasma Pneumoniae (IgG positive) were negative for the presence of IgM antibodies, however Parvovirus was IgM positive.
Clinical course
During hospitalization the patient received two red cell concentrate transfusions along with two furosemide formulas on day 1 due to symptomatic anemia, and on day 6 due to persistence of low Hb levels (5.5 g/dl). Platelet count decreased until day 4 (minimum 84 × 109/L) and WBC count reached minimum levels on day 2, with a 2 × 109/L count (680/L neutrophils and 884/L lymphocytes, reference values of 1900-7500/L and 1000-4800/L respectively).
Symptomatic relief was provided for fever, which resolved on day 4. The patient was discharged on day 12 of hospitalization and followed outpatient care. Haemoglobin reached near-normal values through hospitalization and the patient steadily recovered with significant symptomatic and analytical improvement (Hb 10.1 g/dl, WBC count 6 × 109/L and platelet count 201 × 109/L 12 days after being discharged).
Biochemical investigation
In order to identify possible metabolic or hematological conditions associated with the SBHS, several additional exams were performed but did not establish a definitive diagnosis.
A biochemical lysosomal study was ordered to detect Niemann-Pick Disease types A and B, which were excluded through the determination of the enzimatic activity of sphyngomielinase (0.36 nmol/h/mg/prot considering a reference interval of 0.39-1.38 nmol/h/mg/prot on leucocytes).
Gangliosidosis GM 1 and Mucopolisacaridosis type IV B (Morquio B disease) were also excluded through the determination of the enzimatic activity of beta-galactosidase (169 nmol/h/mg/prot considering a reference interval of 73-585 nmol/h/mg/prot on leucocytes and 26.7 nmol/h/mg/plasma considering a reference interval of 3.6-55 nmol/h/mg/plasma on the plasma).
Determination of the enzimatic activity of beta-glicosidase (6.71 pmol/h/punction considering reference of 1.82-5.99 pmol/h/punction) and of beta-d-chitotriosidase (6.94 pmol/h/punction considering reference 0.773-11.6 pmol/h/punction) both, obtained from dry blood, excluded the possibility of Gaucher Disease.
Plasma beta-d-chitotriosidase and Acid Phosphatase resistant to tartrate were slightly elevated (respectively 114 nmol/h/ml/plasma considering reference 10-85 nmol/h/ml/plasma and 916 nmol/h/ml/plasma considering reference 54-815 nmol/h/ml/plasma), which did not rule out the possibility of Acid Phosphatemia.
Discussion
During the course of the anemia investigation, bone marrow biopsy was performed and revealed a Sea Blue Histiocytosis.
Sea-blue histiocytosis etiology
Several reports indicate that SBHS is associated with metabolic and haematological disorders. Most cases of non-idiopathic SBHS are secondary to lipid metabolic and ceroid storage diseases [11], such as Niemann-Pick Disease [12], lecithin cholesterol acyltransferase deficiency [13,14], Fabry Disease, Gaucher Disease [15] and in association with hyperlipidemia [12], hypercholesterolemia [16], sphingomyelinase deficiency [17], hepatic porphyria [18], hyperlipoproteinemia [19-23], hyperlipoproteinemia type I [20], type III [19], type IV [22], type V [21] and type IIb [24]. In fact, Niemann-Pick Disease type F subgroup, which is seen in adults, has also been termed "sea-blue histiocytosis" [25]. Another subset of diseases that may accompany SBHS are related to high rates of intramedullary cell death, such as chronic myelogenous leukemia [26,27], severe auto-immune neutropenia and idiopathic thrombocytopenic purpura [28-33] or, less frequently, haematological conditions such as thalassemia [34], sickle-cell anemia [7], erythemic myelosis [35], mycosis fungoides [36,37], multiple myeloma and Hodgkin's disease [38]. Certain infections and dermatological conditions have also been linked to this syndrome, namely lepromatous leprosy, leishmaniasis [39] and infectious mononucleosis [18].
A miscellaneous group of diseases have also been sporadically associated with the syndrome, including Batten's disease [40], neuroaxonal dystrophy [37], Takayasu's arteritis [41] and posterior column dysfunction [42,43]. Interestingly, it has been shown that long-term parenteral nutrition with fat emulsion also leads to the development of sea-blue histiocytes in the bone marrow [44]. More recently, it has also been associated with prolonged therapy with liposomal amphotericin B [45]. A summary of possible etiologies of SBHS described here are listed in Table 1.
![]()
Table 1: Summary of Sea-Blue Histiocyte Syndrome etiologies
View Table 1
Conversely, several authors have not been able to identify a possible cause after thorough investigation, thus suggesting the possibility of a primary SBHS [6,8,11,12,28]. These patients where no apparent underlying disorder has been identified are generally below age 40 when diagnosed, do not have a clear family history and the syndrome is often accompanied by hepatosplenomegaly and thrombocytopenia with a mild chronic course [12].
Sea-blue histiocytosis pathophysiological mechanisms
Although a definite mechanism that leads to the development of sea-blue histiocytes has not been established, there is general consensus regarding its lipid and glycolipid nature. This feature is probably associated with abnormal lipid metabolism by histiocytes. One postulate states that this finding might be a secondary phenomenon related to excessive turnover of haematopoietic cells [6,7,28] which promotes an increase in macrophage activity and leads to accumulation of abnormal amounts of cell debris and/or serum lipids [46,47], subsequent storage of oxidation products and macrophage inability to completely metabolize the lipids of phagocytized cells. In fact, most cases in which SBHS occurred were characterized by hypercellularity of the bone marrow [6].
A second postulate suggests that a decrease in sphingomyelinase or a similar enzyme that catalyzes degradation of certain glycosphingolipids produced in the organism might be involved in the formation of these cells [48].
However, several cases have been reported without associated diseases, suggesting the existence of a primary SBHS [6,8,11,12,28]. In this respect, Rywlin and associates [49] suggest that the Sea-blue Histiocyte Syndrome might represent a distinct entity which they termed "idiopathic ceroid histiocytosis of spleen and bone marrow". Still, there is controversy on this subject and no confirmation of a distinct entity for SBHS has been fully established thus far.
Sea-blue histiocytosis manifestations
The most consistent clinical finding of the primary syndrome is splenomegaly [1,12,50], associated with hepatomegaly in approximately 60% of the cases [51], as well as thrombocytopenia accompanied by hemorrhagic diathesis due to bone marrow involvement [12,37,46,52]. Bleeding manifestations such as purpura, epistaxis and massive gastrointestinal hemorrhage are indeed very frequent initial presentations of the disease [12,46,53]. Other associated clinical features have included lymphadenopathy, ophthalmologic abnormalities [12,52,54], pulmonary fibrosis [10,12,54,55], mental retardation [12,52,56] and skin pigmentation [12,52,56,57]. Some patients present with neurological abnormalities in the early stages of the syndrome and may experience ataxia, dementia and seizures several years later [1]. Cutaneous changes are uncommon and include facial macular brownish hyperpigmentation and nodular lesions on the face, trunk, hands and feet [37]. Eyelid infiltration and facial waxy plaques are the most prominent cutaneous feature, resulting in a puffy appearance [37].
Clinical course is generally benign, with a mild chronic progression. Clinical events appear to be associated with the degree of histiocytic involvement of the organs [58] and it has been determined that roughly 15% of cases develop fatal liver failure [1].
No treatment is known except for the associated underlying disorders, which may lead to the resolution of this condition. In most cases where there is no apparent cause, the course of the disease is generally chronic and stable and the prognosis is good [12].
Sea-blue histiocytosis histological features
Regarding the histological features of SBHS, it is characterized by the presence of large (20 μm to 60 μm in diameter) lipid-laden macrophages [41] with simple generic nucleus, and a cytoplasm filled with deep-blue or blue-green granules when stained with Romanovsky or May-Grunwald Giemsa stain [1,41,51,59]. These granules present as round, dense bodies, membranous or lamellated structures, occasionally showing fingerprint appearance and bodies with dense rod-like formations [37]. The mechanism underlying blue granule storage is unknown. They appear yellow-brown with haematoxylin and eosin staining and dark blue with toluidine or giemsa staining. They also show yellow autofluorescence and are highly birefringent under polarized light [37,59].
Histochemical investigations have yet to identify the content of the granules, leading to different theories according to many authors, although there is general consensus that they are composed by glycolipids and phospholipids [60]. In fact, some authors suggest that the storage substance is in fact a glycolipid or phospholipid [1,61], while others presume that they are made of ceroid [21]. The granules may also present with typical ultrastructural features that distinguishes them from metabolic disorders such as Secondary Hemolipidosis, Gaucher's Disease, Niemann-Pick Disease and Ceroid Pigment [60] when these are not associated with SBHS, although there may exist some variability in the degree of cytoplasmic granulation [1,6]. Interestingly, Howard et al. [6] described storage cells with intermediate features between classic sea blue histiocytes and pseudo-gaucher cells in studying a chronic myelogenous leukemia case [6].
Case report discussion and proposed algorithm for patient work-up
As previously mentioned this patients' SBHS was diagnosed during bone marrow examination in the course of a hemolytic anemia associated with hepatosplenomegaly. Taking into account the clinical presentation of this patient, we started investigation of main hematological conditions known to be associated with SBHS.
Chronic myelogenous leukemia and Hodgkin's disease, as well as infectious diseases with hematological involvement were ruled out. During evaluation of blood smear spherocytosis was discovered, a finding that has not been previously associated with SBHS and is seemingly unrelated to the condition.
Further testing was performed, now considering lipid and ceroid storage diseases, which were plausible considering the patient's age group. Niemann-Pick Disease types A and B, Gangliosidosis GM 1 and Mucopolisacaridosis type IV B (Morquio B disease), Gaucher Disease and Acid Phosphatemia enzymatic tests were negative, thus excluding these conditions. Biochemical evaluation also did not raise suspicion of a hyperlipidemic state. Less frequent conditions described in the literature, such as neurological disorders, were not tested since there were no suggestive clinical manifestations. Given that no known or related conditions were discovered during thorough investigation of potential causes, our patient was diagnosed with primary Sea-Blue Histiocyte Syndrome.
The finding of sea-blue histiocytes in the bone-marrow is frequently accidental and associated with investigation of suspected hematological conditions in a patient presenting with related signs and symptoms, as was the case with our patient. However, sea-blue histiocytes may be associated with other unrelated diseases, which may have an impact on patient treatment and prognosis.
In Diagram 1, we propose a preliminary algorithm for patient work-up in the presence of sea-blue histiocytes in the bone marrow. Following these findings, the clinician should be prompted to search for hematological conditions and the presence of lipid/ceroid metabolic disturbance. If the most frequent or suspected conditions have been ruled out, revision of patient work-up including anamnesis, biochemical evaluation and additional tests should be carried out and broadening the spectrum of possible diagnoses with repeat examination of previous tests is recommended. Less frequent conditions such as neurological disorders, adverse effects of medications and rare genetic diseases should be taken into account. If no potential cause to the present condition is found, it can be assumed that the patient suffers from a Primary SBHS. However, a potentially new and undocumented cause for SBHS cannot be ruled out entirely, since the pathophysiological mechanisms and the true nature of the "sea-blue" granules is still unknown. Awareness for this syndrome on behalf of pathologists and clinicians is necessary to prompt the detection of new cases and related causes that might provide further insights on the subject.
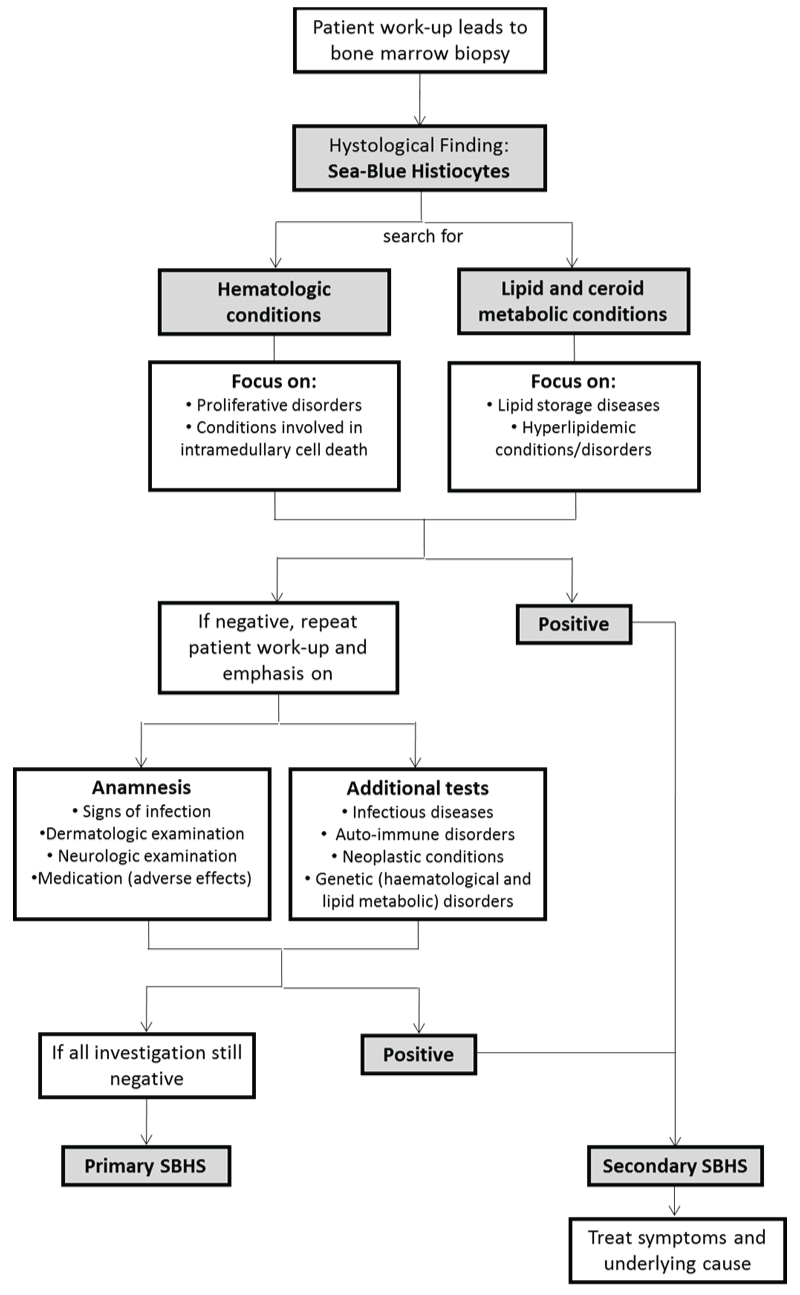
.
Diagram 1: Proposed algorithm for diagnosis of a Sea-Blue Histiocyte Syndrome- Following the findings of sea-blue histiocytes in the bone marrow, the clinician should be prompted to search for hematological conditions and the presence of lipid/ceroid metabolic disturbance by repeating previous exams or performing more specific ones (e.g. enzymatic activity testing). If the most frequent or suspected conditions have been ruled out, revision of patient work-up including anamnesis, biochemical evaluation and additional tests should be carried out and broadening the spectrum of possible diagnosis is recommended. Less frequent conditions such as neurological disorders, adverse effects of medications and rare genetic diseases should be taken into account and investigated if there are suggestive manifestations. If no potential cause to the present condition is found the diagnosis of a Primary SBHS can be made.
View Diagram 1
Authorship contributions
A.P.C. and I.F. did the bibliographic research and review and wrote the manuscript; A.F. was the patient physician and was involved in the hypothesis formulation and clinical diagnosis as well as manuscript revision, F.T. and C.F. developed the hypothesis and were responsible for the histopathologic diagnosis and manuscript revision.
Conflict of interest disclosures
The authors declare no conflicting financial interests.
References
-
Silverstein MN, Ellefson RD, Ahern EJ (1970) The syndrome of the sea-blue histiocyte. N Engl J Med 282: 1-4.
-
Moeschlin S (1947) Die Milzpunktion: Technik, Diagnostische und Hanatikigische. Ergbnisse. 37-38
-
WEWALKA F (1950) [Problem of the blue-pigment macrophages in the sternal punctate]. Wien Klin Wochenschr 62: 788-791.
-
Oo TH, Aish LS, Hassoun H (2002) Unusual presentations of Lymphoma: Case 1: Sea-Blue Histiocytes in Non-Hodgkin's Lymphoma. J Clin Oncol 20: 1942-1943.
-
Bennett JM, Catovsky D, Daniel MT, et al. (1982) Proposals for classification of the myelodysplastic syndrome. Br J Haematol 51:189-199
-
Howard MR, Kesteven PJ (1993) Sea blue histiocytosis: a common abnormality of the bone marrow in myelodysplastic syndromes. J Clin Pathol 46: 1030-1032.
-
Kattlove HE, Gaynor E, Spivack M, Gottfried EI (1970) Sea-blue indigestion. N Engl J Med 282: 630-631.
-
Yamauchi K, Shimamura K (1995) Pulmonary fibrosis and Sea Blue Histiocyte infiltration in a patient with primary myelofibrosis. Eur Respir J 8: 1620-1623.
-
Landas S, Foucar K, Sando GN, Ellefson R, Hamilton HE (1985) Adult Niemann-Pick disease masquerading as sea blue histiocyte syndrome: report of a case confirmed by lipid analysis and enzyme assays. Am J Hematol 20: 391-400.
-
Sawitsky A, Hyman GA, Hyman JB (1954) An unidentified reticuloendothelial cell in bone marrow and spleen; report of two cases with histochemical studies. Blood 9: 977-985.
-
Suzuki O, Abe M (2007) Secondary Sea Blue Histiocytosis derived from Niemann Pick Disease. J Clin Exp Hematop 47:19-21.
-
Sawitsky A, Rosner F, Chodsky S (1972) The sea-blue histiocyte syndrome, a review: genetic and biochemical studies. Semin Hematol 9: 285-297.
-
Naghashpour M, Cualing H (2009) Splenomegaly with sea-blue histiocytosis, dysplipidemia, and nephropathy in a patient with lecthin-cholesterol acyltransferase deficiency: a clinicopathologic correlation. Metabolism 58: 1459-1464.
-
Jacobsen CD, Gjone E, Hovig T (1972) Sea-blue histiocytes in familial lecithin: cholesterol acyltransferase deficiency. Scand J Haematol 9: 106-113.
-
Baumgartner C, Bucher U (1975) Blaue Pigmentmakrophagen (sea blue histiocytes) and Gaucher-aehnliche Zellen. Vorkommen und Bedeutung Blut 30:309.
-
Rosen Y, Lin HS, Peress NS (1974) Alcoholism, hypercholesterolemia, and reticuloendothelial histiocytic lipidosis. Postmortem study. N Y State J Med 74: 851-854.
-
Konagaya M, Konishi T, Konagaya Y, et al. (1989) Partial sphingomyelinase deficiency with sea blue histiocytosis and neurovisceral dysfunction. Jpn J Med 28: 85-88.
-
Ghosh ML (1972) The sea-blue histiocyte syndrome with hepatic porphyria and infectious mononucleosis. J Clin Pathol 25: 945-946.
-
Fredrickson DS, Lees RS (1965) A system for phenotyping hyperlipoproteinemia. Circulation 31: 321-327.
-
Ferrans VJ, Buja LM, Roberts WC, et al. (1971) The spleen in type I hyperlipoproteinemia. Histochemical, biochemical, microfluorometric and electron microscopic observations. Am J Pathol 64: 67-96.
-
Rywlin AM, Lopez-Gomez A, Tachmes P, et al. (1971) Ceroid histiocytosis of the spleen in hyperlipidemia: relationship to the syndrome of the sea-blue histiocyte. Am J Clin Pathol 56: 572-579.
-
Castoldi G, Grusovin GD, Scapoli G (1974) Sea-blue histiocytosis in hyperlipoproteinemia (cytomorphological, clinical and pathogenetic aspects). Haematologica 59: 68-80.
-
Parker AC, Bain AD, Brydon WG, Harkness RA, Smith AF, et al. (1976) Sea-blue histiocytosis associated with hyperlipidaemia. J Clin Pathol 29: 634-638.
-
Lee YB, Kim H, Park CI, et al. (1983) Sea-blue histiocytosis associated with hyperlipoproteinemia type IIb. Yonsei Med J 24: 132-140
-
Günay E, Firrat Güven S, Aktas Z, Sipit T, Agackiran Y, et al., (2012) Pulmonary involvement in sea-blue histiocytosis. Tuberk Toraks 60: 176-179.
-
Cazal P, Izarn P, Emberger JM, Navarro M, Rizkalla N (1971) [Splenomegaly with ceroids (blue histiocyte syndrome)]. Ann Anat Pathol (Paris) 16: 405-416.
-
Dosik H, Rosner F, Sawitsky A (1972) Acquired lipidosis: Gaucher-like cells and "blue cells" in chronic granulocytic leukemia. Semin Hematol 9: 309-316.
-
Ganguly S, Cunningham MT (2004) Idiopathic thrombocytopenic purpura associated with bone marrow sea-blue histiocytosis. Am J Hematol 77: 405-406.
-
Richards DM (1989) Disorders of the Spleen: Barbara C. Wolf MD, Richard S. Neiman MD. W B Sauders Company. Blood Reviews 3: 277.
-
Cohn J, Tygstrup I (1976) Foamy histiocytosis of the spleen in patients with chronic thrombocytopenia. Scand J Haematol 16: 33-37.
-
Ishihara T, Matsumoto N, Uchino F (1974) Foamy histiocytes in the spleen associated with idiopathic thrombocytopenic purpura. Acta Pathol Jpn 24: 273-284.
-
Quattrin N, Di Girolamo R, Mastrobuoni A, Montuori R (1972) [New hemorrhagic thrombocytopenic hemorrhagic disease: sea-blue histiocytosis]. Minerva Med 63: 1396-1404.
-
Summerell JM, Gibbs WN (1972) Splenic histiocytosis associated with thrombocytopenia. Acta Haematol 48: 34-38.
-
Beltrami CA, Bearzi I, Fabris G (1973) Storage cells of spleen and bone marrow in thalassemia: an ultrastructural study. Blood 41: 901-912.
-
Karayalcin G, Rosner F, Sawitsky A (1971) Sea-blue histiocyte syndrome in an octagenarian [letter]. Lancet 2: 318.
-
Robinowitz B, Roenigk HH Jr, Smith MM (1975) Sea-blue histiocytes in mycosis fungoides. Arch Dermatol 111: 1165-1167.
-
Caputo R (1998) Text Atlas of Histiocytic Syndromes: A Dermatological Perspective. London, UK. Martin Dunitz.
-
Chandra P, Rosner F, Sawitsky A (1973) Letter: Sea-blue histiocytes in thrombocytopenic purpura. Ann Intern Med 79: 901-902.
-
Safatle-Ribeiro AV, Baba ER, Iriya K, Fylyk SN, Pennacchi C, et al. (2010) Double-balloon endoscopy reveals sea-blue histiocytosis affecting the small bowel (with video). Gastrointest Endosc 72: 1266.
-
Smith H (1974) Sea-blue histiocytes in marrow in Batten-Spielmeyer-Vogt disease. Pathology 6: 323-327.
-
Tadmor R, Aghai E, Sarova-Pinhas I, Braham J (1976) Sea-blue histiocytes in a case of Takayasu arteritis. JAMA 235: 2852-2853.
-
Swaiman KF, Halverson D, Sharp HL, Brunning RD, Lockman LA (1973) Sea-blue histiocyte associated with posterior column dysfunction in childhood. Neurology 23: 474-477.
-
Swaiman KF, Garg BP, Lockman LA (1975) Sea-blue histiocyte and posterior column dysfunction: a familial disorder. Neurology 25: 1084-1087.
-
Bigorgne C, Le Tourneau A, Messing B, et al. (1996) Sea-blue histiocyte syndrome in bone marrow secondary to total parental nutrition including fat-emulsion sources: a clinicopathologic study of seven cases; Br J Haematol 95: 258-262.
-
Michot JM, Gubavu C, Fourn E, et al. (2014) Very prolonged liposomal amphotericin B use leading to a lysosomal storage disease. Int J Antimicrob Agents 43: 566-569.
-
Román Irizarry LA, Barquet-Chediak A (1968) Reticuloendotheliosis with blue-staining granules. A disease resembling Niemann-Pick's with Sjogren's Syndrome. Bol Asoc Med P R 60: 230-237.
-
Levine SB, Stone WH, Fitzwater JE, et al. (1974) Sea-blue histiocytes [letter]. Archives of Pathology 98: 283.
-
Golde DW, Schneider EL, Bainton DF, Pentchev PG, Brady RO, et al. (1975) Pathogenesis of one variant of sea-blue histiocytosis. Lab Invest 33: 371-378.
-
Rywlin AM, Hernandez JA, Chastain DE, Pardo V (1971) Ceroid histiocytosis of spleen and bone marrow in idiopathic thrombocytopenic purpura (ITP): a contribution to the understanding of the sea-blue histiocyte. Blood 37: 587-593.
-
Silverstein MN, Ellefson RD (1972) The syndrome of the sea-blue histiocyte. Semin Hematol 9: 293-307.
-
Varela-Duran J, Roholt PC, Ratliff NB Jr (1980) Sea-blue histiocyte syndrome. A secondary degenerative process of macrophages? Arch Pathol Lab Med 104: 30-34.
-
Saidi P, Azizi SP, Sarlati R, Sayar N (1970) Rare variant of lipid storage disorders. Blood 35: 533-538.
-
Marmont A, Santini G (1975) [Clinico-hematological aspects of a case of sea-blue histiocytosis in an adult]. Minerva Med 66: 2458-2460.
-
Cogan DG, Federman DD (1964) Retinal involvement with reticuloendotheliosis of unclassified type. Arch Ophtalmol 71: 489-491.
-
Hermansky F, Pudlak P (1959) Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: report of two cases with histochemical studies. Blood 14: 162-169.
-
Lake BD, Stephens R, Neville BG (1970) Syndrome of the sea-blue histiocyte. Lancet 2: 309.
-
MALININ TI (1961) Unidentified reticuloendothelial cell storage disease. Blood 17: 675-686.
-
Fadilah SA, Mazeni NR, Cheong SK (2000) Successful pregnancy in a patient with familial sea-blue histiocyte syndrome. Med J Malaysia 55: 510-512.
-
Zina AM, Bundino S, Pippione M (1987) Sea-blue histiocyte syndrome with cutaneous involvement. Case report with ultrastructural findings. Dermatologica 174: 39-44.
-
Reidbord HR, Horvat BL, Fisher ER (1972) Splenic Lipidoses: Histochemical and Ultrastructural Differentiation with special reference to the syndrome of the Sea-Blue Histiocyte. Arch Pathol 93: 518-524.
-
Silverstein MN, Young DG, Remine WH, Pease GL (1964) Splenomegaly With Rare Morphologically Distinct Histiocyte; A Syndrome. Arch Intern Med 114: 251-257.
