

International Journal of Respiratory and Pulmonary Medicine
Genetics and its Associated Pathways of Pulmonary Arterial Hypertension
Rui Fan1,2, Daniel Penny1 and Yuxin Fan1*
1Department of Pediatrics, Texas Children's Hospital, Baylor College of Medicine, USA
2Department of Pediatrics, Xijing Hospital, The Fourth Military Medical University, China
*Corresponding author: Yuxin Fan, M.D., Ph.D., John Welsh Cardiovascular Diagnostic Laboratory, Section of Cardiology, Department of Pediatrics, Texas Children's Hospital, Baylor College of Medicine, 1102 Bates Ave, Suite 430.09, Houston, Texas 77030, USA, Tel: 832-824-4155, Fax: 832-825-5159, Email: yuxinf@bcm.edu
Int J Respir Pulm Med, IJRPM-2-031, (Volume 2, Issue 4), Review article; ISSN: 2378-3516
Received: September 04, 2015 | Accepted: October 20, 2015 | Published: October 23, 2015
Citation: Fan R, Penny D, Fan Y (2015) Genetics and its Associated Pathways of Pulmonary Arterial Hypertension. Int J Respir Pulm Med 2:031. 10.23937/2378-3516/1410031
Copyright: © 2015 Fan R, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Pulmonary arterial hypertension (PAH) is a devastating disease with significantly reduced survival. To date, no tested therapies have demonstrated an ability to reverse or cure the disease. The etiology of PAH is heterogeneous and incompletely understood. Genetics plays an important role in idiopathic and heritable PAH. Many germline gene mutations have been described as promoters of PAH and include mutations in the gene coding BMPR2, which has been identified in at least 50% of familial and 10-40% of sporadic cases. Mutations in ACVRL1 (ALK-1), endoglin (ENG), SMAD4 and SMAD8, and other TGF-β family members are additional rare causes of PAH. CAV 1 regulates SMAD2/3 phosphorylation, and mutations in CAV1 are a rare cause of PAH, KCNK3 and KCNK5 are members of potassium channels expressed in pulmonary artery smooth muscle cells, and mutations in them are rare causes of both heritable and idiopathic PAH. According to Notch signaling pathway, mutations in NOTCH3 in PAH patients with PAH showed that these mutations were involved in cell proliferation and viability. Heterozygous variants in NOTCH1 are an additional cause of Adams-Oliver syndrome in many families. EIF2AK4 has been identified as the major gene linked to pulmonary veno-occlusive disease. Recent advanced DNA sequencing technologies have facilitated the discovery of additional genes with mutations. Genetic testing is also available for PAH and should be considered in families who are at increased risk of developing PAH. Ultimately, a profound understanding of the genetic factors relevant to PAH will further our understanding and knowledge to promote novel therapeutic development.
Keywords
Pulmonary arterial hypertension (PAH), Genetics, Next generation sequencing (NGS), Whole exome sequencing (WES), Whole genome sequencing (WGS)
Abbreviations
PAH: Pulmonary Arterial Hypertension, PAP: Pulmonary Artery Pressure, PCWP: Pulmonary Capillary Wedge Pressure, WSPH: World Symposium on Pulmonary Hypertension, PVR: Pulmonary Vascular Resistance, HPAH: Heritable PAH, IPAH: Idiopathic PAH, HIV: Human Immunodeficiency Virus, NGS: Next Generation Sequencing, WES: Whole Exome Sequencing, BMPR2: Bone Morphogenetic Protein Receptor Type II, TGF-β: Transforming Growth Factor, BMPS: Bone Morphogenetic Proteins, HLMVE: Human Lung Microvascular Endothelial, SARA: Smad Anchor for Receptor Activation, HHT: Hereditary Hemorrhagic Telangiectasia, ENG: Endoglin, eNOS: endothelial Nitric Oxide Synthase, SNVs: Single Nucleotide Variants, PASMC: Pulmonary Artery Smooth Muscle Cells, eIF2α: eukaryotic Initiation Factor 2, TGFBR2: TGF-β receptor-2, AOS: Adams-Oliver Syndrome
Introduction
Pulmonary arterial hypertension (PAH) is a rare disease characterized by a sustained increase in pulmonary artery pressure (PAP), without common causes of pulmonary hypertension such as heart, lung, and other chronic diseases [1]. PAH is a clinical diagnosis that cannot be made accurately without hemodynamic measurements taken from pulmonary artery catheterization. The current consensus regarding hemodynamic criteria of PAH include mean PAP (mPAP) of 25 mm Hg or more and pulmonary capillary wedge pressure (PCWP) of 15 mm Hg or less [2]. In the current United States guidelines, according to the 5th World Symposium on Pulmonary Hypertension (WSPH) held in 2013 in Nice, pulmonary vascular resistance (PVR) > 3 wood unit (WU) is added as part of the hemodynamic definition of PAH [3-6].
The clinical classification of PAH has been adopted worldwide and recently was updated. According to this system, Group 1 PAH is divided into disease subgroups including heritable (HPAH, formerly familial PAH), idiopathic (IPAH), and PAH associated with drug/toxin exposures or a variety of other medical conditions including connective tissue diseases, human immunodeficiency virus (HIV) infection, congenital heart disease, and portal hypertension [7,8].
PAH is pathologically characterized by progressive intimal proliferation, smooth muscle cell hypertrophy, and surrounding adventitial expansion leading to occlusive vascular lesions of the small pulmonary arteries [9]. The pathogenesis of PAH is complex, including both genetic and environmental factors altering vascular structure and functions, and incompletely understood [10]. Despite advancements in therapy made during the past 25 years, PAH remains a devastating disease with significantly reduced survival rates [11]. No therapies tested to date have demonstrated an ability to reverse or cure PAH [12]. Further understanding of the pathophysiological mechanisms underlying PAH to promote novel therapeutic development [13,14] is profoundly needed. The identification of PAH genes, such as bone morphogenetic protein receptor type II (BMPR2), and the recognition of critical path biological abnormalities associated with them, now provide a unique opportunity to better understand the disease.
We herein provide a review of the literature and describe the genetics and its associated pathways of PAH.
Genetics of PAH
Genetic approaches and genes associated with PAH
Familial cases of PAH have been long recognized before the discovery of genetic mutations associated with HPAH and are usually autosomal dominantly inherited. Of the cases previously thought to be IPAH, 10 - 40% have identifiable mutations in their BMPR2 and, therefore, pose a hereditary risk for other family members. However, a family history of PAH may go unrecognized in IPAH cases with BMPR2 mutations, due to undiagnosed diseases, incomplete penetrance, or spontaneous mutations [15].
Various strategies have been applied to identify the PAH associated genes. Initially large families were required for linkage analysis which mapped PPH1 to chromosome 2q31 - q32 [16]. Positional candidate genes at 2q21 - 32 were sequenced, and BMPR2 was firstly identified as the major genetic cause of PAH [17]. Next-generation sequencing (NGS) technologies made possible the discovery of unknown PAH genes and are capable of screening the entire genome for genetic variants relevant to the pathogenesis of common and Mendelian disorders [18]. In particular, whole exome sequencing (WES) has been successfully applied to identify novel candidate genes in hereditary disorders [19,20]. Since 2000, mutations in other genes related to BMPR2 signaling, such as mutations in ALK1, ENG and SMAD8, have been discovered, and progress has been made to identify genetic and epigenetic modifiers of disease expression. More recently, use of WES in patients with BMPR2-negative familial PAH has identified CAV-1 and KCNK3 as two new candidate genes that may increase susceptibility for PAH in carriers (Table 1) [21,22]. With the advent of WES, rarer monogenic causes of PAH have been identified, using smaller families than required for linkage [21]. In addition, a genome-wide association study (GWAS) has successfully identified common Variants predisposing to PAH [23].
![]()
Table 1: Genes Associated with PAH
View Table 1
Mutations in TGF-β/BMP signaling pathway
Bone morphogenetic protein receptor type II (BMPR2) is a member of the transforming growth factor (TGF-β) superfamily of transmembrane serine/threonine kinase receptors. It binds bone morphogenetic proteins (BMPs), which are involved in a series of cellular functions including osteogenesis and cell growth and differentiation. Signaling in the BMP pathway begins with the binding of a BMP to the type II receptor, which causes the recruitment of a BMP type I receptor, and phosphorylates a receptor-regulated Smad (R-Smad) transcriptional regulator (Figure 1) [24].
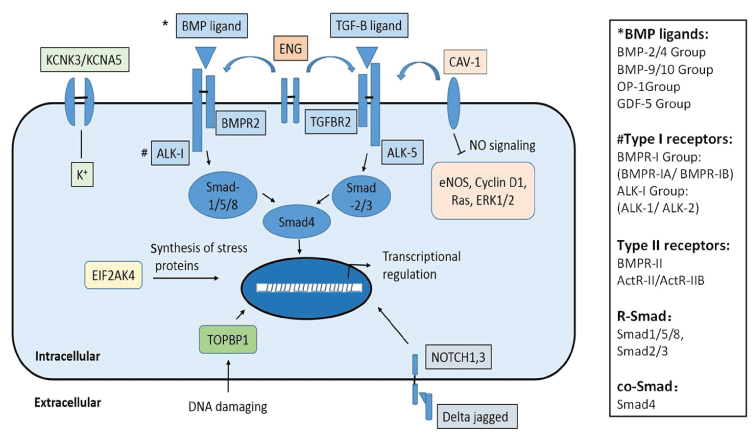
.
Figure 1: Genes with mutations known to associate with PAH or PAH-related diseases
Except for genes involved in TGF-β/BMP signaling pathway including BMPR2, ACVRL1, END, SMAD9 (encodes SMAD 8), SMAD4, and BMP9, there are several other genes and cell signaling pathways that contribute to PAH: NO signaling and eNOS-derived oxidative stress pathway: CAV1; DNA damage and replication stress response: TOPBP1; The potassium channel-related genes: KCNK3, KCNA5; Notch signaling pathway: NOTCH1, NOTCH3; and Autosomal recessive HPAH related gene EIF2AK4. Possible resultant signaling or effects of protein actions are briefly listed. And see column on the right side for BMP sub-groups, type II and type I receptors, and Smad proteins in TGF-β/BMP signal transduction.
View Figure 1
The main function of BMPR2 is inhibiting the proliferation of vascular smooth muscle cells. Alteration of this gene leads to vascular smooth muscle proliferation and then pulmonary hypertension [25]. In the 1990s, two teams working independently on a single gene that might be responsible for the majority of HPAH cases identified by different methods BMPR2 as the target gene [26,27]. Since these initial reports, more than 400 different mutations on BMPR2 gene definitively associated with HPAH have been detected using diverse approaches such as direct sequencing, Southern blotting, and multiplex ligation-dependent probe amplification [28]. Germ line mutations in BMPR2 were identified in at least 50% of heritable and 40% of sporadic cases [27,29]. Approximately 30% of them are missense mutations that occur in highly conserved amino acids and are likely to perturb ligand-receptor binding or disrupt the constitutively active functional domains of the receptor. Most of the coding mutations are frameshift and nonsense mutations or involve deletions [30]. Many of the mutations were predicted to cause premature truncation that might trigger nonsense-mediated decay of the mutant mRNA and lead to a state of haploinsufficiency, which may represent the predominant molecular mechanism underlying inherited predisposition to PAH [31]. Previous studies have also shown that patients with a BMPR2 mutation have worse prognoses than those of non-carriers [32,33]. BMPR2 mutations are especially important and require screening of the relatives of patients with heritable or idiopathic pulmonary hypertension. Studies also have found a correlation between BMPR2 and exercise-induced elevation of PAP [34].
Whether the type of BMPR2 mutation (with truncating or missense) alters PAH prognosis remains controversial [35,36]. Wang et al. compared the effects of two missense mutations (Tyr67Cys ECD and Ser863AsnCD) and two truncating mutations (Thr268fs and Gln433X in KD) on the release of nitric oxide (NO) and endothelin-1 (ET-1) and on endothelial cell proliferation, migration, and apoptosis. The results demonstrated that only the Ser863Asp mutant stimulates production of ET-1 by human lung microvascular endothelial (HLMVE) cells. The increased level of ET-1 associated with the Ser863Asp mutant and the decreased levels of NO in all four mutants may represent a mechanism that explains how BMPR2 mutations are associated with the development of PAH [37]. This is the first study to compare the functional significance of missense and truncating mutations of BMPR2.
Taken together with previously reported data, more than 400 mutations of BMPR2 have been found, and the majority of them located in exons encoding the ligand-binding domain (exons 2-3) and key catalytic regions of the kinase domain (exons 6-9,11). While exons 1, 4, 10 and 13, encoding receptor regions of uncertain importance to function, have a low frequency of missense mutation. Cysteine substitutions comprise the majority of missense mutations in the extracellular ligand-binding domain and are concentrated on 9 of 10 conserved residues, which are essential for the integrity of its three-dimensional structure [38,39]. Girerd et al. compared the age at diagnosis, severity, and prognosis of 171 PAH patients whose mutations located in the cytoplasmic tail of BMPR2 with these located on the signal peptide, the ligand domain, or the kinase domain. They found that patients carrying a mutation in the cytoplasmic tail were characterized by older ages at diagnosis, less severe hemodynamic characteristics, and a greater chance of being a long-term responder to calcium channel blockers. In vitro experiments showed that mutations located in the cytoplasmic tail led to normal activation of the Smad pathway, whereas activation was abolished in the presence of mutations located in the kinase domain [40]. Further investigations are needed to better understand the consequences of these BMPR2 mutations in TGF-β signaling pathway.
Smad-dependent signaling: Smad4, Smad8/9
Smad4 is involved in many cell functions such as cell differentiation, apoptosis, and embryonic development. It modulates members of the TGF-β protein superfamily by binding R-Smads and then forms a complex that binds to DNA and serves as a transcription factor [41]. Smad8, also known as Smad9, is a receptor regulated Smad (R-Smad) [42]. BMPR2 triggers signal transduction through the R-Smads (Smad1, Smad5, and Smad8) upon ligand binding and complex formation with a type I receptor, namely BMPR-1A, -1B, or ACVRL1. When a BMP binds to a receptor, it causes Smad8 to interact with Smad anchor for receptor activation (SARA). The binding results in the phosphorylation of the Smad8 protein, dissociation from SARA, and association with Smad4. R-Smads translocate to the nucleus in complex with co-Smad4 to regulate transcription of target genes [43].
Previous reports suggested that the loss of signals mediated by Smad1/5/8 plays an important role in pulmonary vascular remodeling and the pathogenesis of PAH [44]. Shintani et al. identified a nonsense mutation in Smad8, c.606C > A (p.C202X), in one patient among 23 patients clinically diagnosed with IPAH and with no mutations in BMPR2 or ACVRL1. Immunoblotting and co-immunoprecipitation assay showed that it was not phosphorylated by TGF-β/BMP type I receptors and that it did not interact with Smad4. These results indicate the Smad8 mutant disturbs the downstream signaling of TGF- β/BMP pathway [45]. Nasim et al. reported two gene defects in Smad4 and Smad8 among a cohort of 324 PAH cases, neither detected in a substantial control population. A putative splice site mutation in SMAD4 resulted in moderate transcript loss due to compromised splicing efficiency. Functional analyses suggest significant, albeit limited, consequences on transcript integrity, signaling, and target gene regulation [46].
HHT related signaling: ACVRL1, ENG, and BMP-9
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant vascular disorder characterized by mucocutaneous telangiectases, recurrent epistaxes, and macroscopic arteriovenous malformations, particularly in the pulmonary, hepatic, and cerebral circulation. Pulmonary arteriovenous malformations may create clinically significant right-to-left shunts, causing hypoxemia, paradoxical embolism, stroke, and cerebral abscesses [47-50]. In patients with HHT, postcapillary pulmonary hypertension may develop as a consequence of a hyperkinetic state, resulting in high cardiac output heart failure. However, HHT is also associated with a precapillary pattern of pulmonary hypertension that is histologically indistinguishable from IPAH [33].
Activin A receptor type II-like kinase-1 (ACVRL1, also known as ALK1) and ENG mutations have been reported as causes of HHT and/or HPAH [51-54]. ACVRL1 is a type I cell-surface receptor for the TGF- β superfamily, sharing with other type I receptors a high similarity in serine-threonine kinase subdomains, a glycine- and serine-rich region (called the GS domain) preceding the kinase domain, and a short C-terminal tail [55]. ACVRL1 acts downstream of the type II receptors and determines the signaling specificity by recruiting R-Smads. In the study undertaken by Girerd et al. ACVRL1 mutation carriers identified from the French PAH Network and from previously published cases of patients with IPAH or with PAH and carrying a BMPR2 mutation were compared for clinical, functional, and hemodynamic characteristics. They demonstrated that age at the time of PAH diagnosis and death of ACVRL1 mutation carriers were significantly lower than those of the noncarriers of the BMPR2/ACVRL1 mutation [28,56,57]. The observation that ACVRL1 mutation carriers develop PAH earlier may illustrate the constitutive susceptibility conferred by the mutation on the course of the disease. The formation of a heteromeric complex with BMPR-II and ACVRL1 has been proposed [58,59]. ACVRL1 haploinsufficiency may lead to a pulmonary vascular status that predisposes to the development of PAH.
ENG is a type III receptor with no kinase activity that enhances ligand binding to its receptors [60]. A germline ENG mutation was identified in a patient with HHT, revealing that both ENG and ACVRL1 can be involved in PAH and HHT. Moreover, patients with ENG mutation seem to be at lower risk of developing PAH compared with carriers of the ACVRL1 mutation [61].
BMPs are the largest subgroup of signaling molecules in the TGF-β superfamily. BMPs play crucial roles during embryonic development and exerts many functional effects, such as cell apoptosis, proliferation, differentiation, migration, and angiogenesis [57], by binding to specific endothelial cell surface receptors, namely ENG and ACVRL1. Next, the BMP9-dependent activation of ACVRL1 leads to the phosphorylation of Smad1, Smad5, and Smad8, and then it associates with Smad4 to form a Smad complex to regulate gene expression [62]. Mutations in BMP9 identified in patients with HHT strongly suggest that alterations in BMP9, the upstream component of this signaling route, cause a vascular-malformation syndrome with phenotypic overlap with HHT, and pathogenic mutations in ENG, ACVRL1 and SMAD4, give rise to HHT1, HHT2, and JP-HHT, respectively (Figure 2) [63,64].
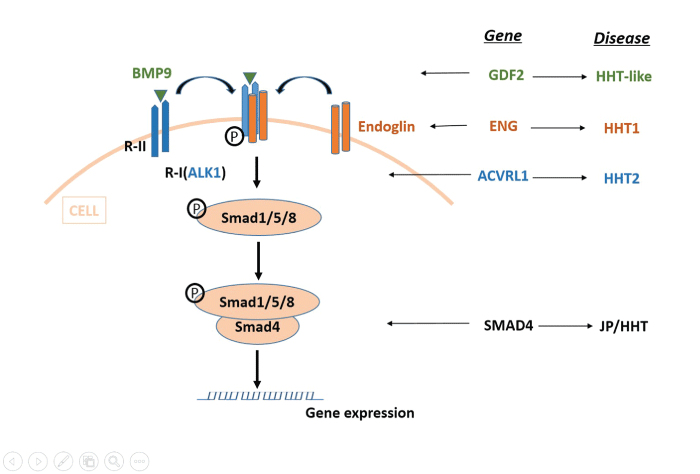
.
Figure 2: BMP9 and the TGF-β signaling pathway
BMP9 binds to specific type I (R-I) and type II (R-II) cell-surface receptors as well as to the auxiliary receptor Endoglin. R-II then phosphorylates ALK1 (R-I), which can induce phosphorylation of receptor-regulated (R-Smads) Smad1/5/8. Once phosphorylated, R-Smads form heteromeric complexes with a cooperating homolog (co-Smad) Smad4, and translocate into the nucleus where they regulate the transcriptional activity of target genes. BMP9 is encoded by GDF2, ALK1 by ACVRL1 and Endoglin by ENG. Mutations in GDF2, ACVRL1, SAMD4 may give rise to HHT1, HHT2, and JP-HHT respectively.
View Figure 2
In PAH there is also an up-regulation of the ET-1 system that has been shown to regulate endothelial cell migration and angiogenesis [65]. Plasma levels of ET-1 correlate positively with haemodynamic severity in patients with PAH and negatively with outcome [66]. Variant studies have shown recently that BMP-9 stimulates ET-1 release in vascular endothelial cells, suggesting that BMP-9 may be a circulating factor affecting angiogenesis and endothelial stabilization. However, this effect depends on BMPR II signaling transduction, argues against an important role for BMP-9 as a source of increased endothelial ET-1 production in human PAH [67,68].
PAH not attributable to mutations in TGF- β genes
NO signalling and eNOS-derived oxidative stress
One of the key initiating events leading to PAH is endothelial dysfunction characterized by uncoupling of endothelial nitric oxide synthase (eNOS) and an associated lack of bioavailable nitric oxide (NO) [69]. Loss of NO signaling and increased eNOS-derived oxidative stress are central to the pathogenesis of PAH [70,71]. Several reported studies showed that dissociation or loss of caveolin-1 (CAV-1) leads to pathological eNOS hyperactivity [72-74]. The expression of CAV1 is necessary for the formation of caveolae [75], which is critical to many receptor signaling cascades relevant to PAH, including the NO pathway [71,76]. Bauer et al. reported finding high-level expression of thrombospondin-1 (TSP1) and CD47 in the lungs of patients with PAH and increased expression of them in experimental models of PAH. Furthermore, the therapeutic blockade of CD47 activation in hypoxic pulmonary artery endothelial cells upregulated CAV-1, increased CAV-1-CD47 co-association, decreased eNOS-derived superoxide and protected animals from developing PAH. Briefly speaking, activated CD47 promotes disease by limiting CAV-1 inhibition of dysregulated eNOS [77]. Austin et al. used WES to evaluate a large family with autosomal dominant HPAH and identified a de novo CAV1 mutation, which then was confirmed in an unrelated patient with PAH. Although originally classified as having IPAH, this patient thus has a heritable form of PAH as well. The association of PAH with CAV1 mutations is the first example of a genetic mutation associated with human PAH not directly involved in TGF-β signaling, and affirms the importance of intact caveolar function to pulmonary vascular homoeostasis in humans [78].
DNA damage and replication stress response
In a previous study, use of WES to screen the genome of 12 patients with IPAH led to the identification of mutations in topoisomerase DNA binding II binding protein 1 (TopBP1), a novel gene known associated with DNA damage response and cell survival. Vinicio, et al. found that pulmonary microvascular endothelial cells (PMVECs) purified from lungs of patients with IPAH contained more TopBP1 single nucleotide variants (SNVs). This finding is relevant, considering that TopBP1 expression is impaired in IPAH PMVECs and correlates with reduced survival. These researchers proposed that TopBP1 is required for the DNA damage response in the setting of injury and its absence may predispose to cell death and impaired angiogenesis [79,80].
The potassium channels: KCNK3, KCNK5
KCNK3 (potassium channel subfamily K, member 3), also called TASK-1, belongs to a family of mammalian potassium (Kv) channels that are characterized by the presence of four transmembrane domains and two pore domains per subunit [81,82]. Ion channels play critical roles in vascular remodeling, and KCNK3 is thought to be involved in the regulation of vascular remodeling and abnormal vascular proliferation in patients with PAH by preventing apoptosis [83]. Ma et al. studied a family in which multiple members had PAH without identifiable mutations in any of the genes known to be associated with the disease, including BMPR2, ACVRL, ENG, SMAD9, and CAV-1. They identified a novel heterozygous missense variant in KCNK3, and five additional heterozygous missense variants in KCNK3 were independently identified in 92 unrelated patients with familial PAH and 230 patients with IPAH. The study identified the association of a novel gene, KCNK3, as a disease-causing candidate gene with HPAH and IPAH. Because KCNK3 channels are not voltage-dependent and are open at negative potentials, these mutations probably cause depolarization of the resting membrane potential, which could lead to pulmonary-artery vasoconstriction [21]. Mutations in this gene produced a reduced Kv-channel current, which was successfully remedied by pharmacologic manipulation [84].
It is also possible that in patients with PAH, variation in KCNK3 function may be a more broadly applicable risk factor that is not caused by mutations in KCNK3. In addition, previous studies of Kv channels support the concept that the expression or function of Kv channels is altered in patients with idiopathic PAH, and dysfunctional Kv-channel activity may contribute to the development or persistence of PAH [85]. In a study of mice with wild-type Kv channels, therapeutic Kv-channel activation was useful in the treatment of established PAH in the absence of known genetic variations in Kv channels [84]. Thus, the therapeutic targeting of KCNK3 may be beneficial for patients with PAH who have increased vascular tone independent of their KCNK3 genetic status.
The pore-forming subunit, Kv1.5, forms functional voltage-gated Kv channels in human pulmonary artery smooth muscle cells (PASMC), and abnormal Kv channel Kv1.5 function encoded by KCNA5 mutations has been reported previously in patients with IPAH [86]. A recent study of our lab was the first to identify digenic mutations, a novel mutation in KCNA5 and a known mutation in BMPR2, supporting a 'two-hit' model for the severe disease course in an 13-year-old African-American girl with severe HPAH. By sequencing the candidate genes, we identified both a heterozygous missense mutation in the BMPR2 gene and a heterozygous deletion mutation in the KCNA5 gene, which results in a frameshift mutation and leads to premature truncation in the C-terminal of the potassium channel Kv1.5. The 'two-hit' of the digenic mutations may account for the early occurrence, severity and rapid deterioration of PAH in this patient. The girl presented for cardiology evaluation at six years of age following a syncopal episode that led to her diagnosis of PAH. Her father had presented with PAH earlier the same year at the age of 25 and died of complications within one year. The family history is otherwise negative. The severe symptoms of PAH and rapid deterioration in her father may be due to the BMPR2 mutant. However, the occurrence is at adult age, much later than the index patient's at presentation. The absence of clinical PAH symptoms in the mother may be due to incomplete penetrance of the KCNA5 mutation and supports the second-hit modifier model as well [87].
Autosomal recessive HPAH: EIFAK4
Pulmonary veno-occlusive disease (PVOD) is a rare and devastating cause of pulmonary hypertension that is characterized histologically by widespread fibrous intimal proliferation of septal veins and preseptal venules. It is frequently associated with pulmonary capillary dilatation and proliferation [88,89]. Using WES, Eyries et al. detected recessive mutations in EIF2AK4 gene in 13 related PVOD families with history of PVOD. Biallelic EIF2AK4 mutations in 5 of 20 histologically confirmed sporadic cases of PVOD had also been reported. All mutations, either in a homozygous or compound-heterozygous state, disrupted the function of this gene. These findings indicated EIF2AK4 as the major gene linked to development of PVOD [90].
EIF2AK4 belongs to a family of four kinases that phosphorylate the α-subunit of the eukaryotic initiation factor 2 (eIF2α). EIF2AK4-mediated phosphorylation of eIF2α inactivates the factor and leads to preferential synthesis of stress proteins. The pathophysiological link between biallelic EIF2AK4 loss-of-function mutations and vascular cell proliferation and remodeling of lung vessels remains elusive. The involvement of EIF2AK4 in PVOD could be related either directly to its amino acid starvation sensor function and subsequent translational changes secondary to its activation or to EIF2AK4 kinase activity, which might have substrates other than eIF2α. Two series of experimental data potentially connect EIF2AK4 to the BMP pathway, which has been implicated in PAH pathogenesis through mutations in BMPR2 and ACVRL1 [91,92]. Notably, EIF2AK4 was also found to interact with SMAD4 and SMAD1 [93] and also with ACVRL1, ENG and TGF-β receptor 2 (TGFBR2). The identification of EIF2AK4 mutations as the major cause of heritable PVOD confirms the hereditary origin of this particular type of pulmonary hypertension and allows identification of heritable but apparently sporadic cases.
Notch signaling pathway: NOTCH1, NOTCH3
NOTCH genes encode a group of single-pass transmembrane receptors (NOTCH1-4). The large extracellular domain contains tandem EGF-like repeats and cysteine-rich Notch/LIN-12 repeats, ankyrin repeats and a PEST sequence have been found within the intracellular domain [94]. The NOTCH signaling pathway is highly conserved and critical for cell fate determination during embryonic development, including many aspects of vascular development [95]. Despite the involvement of the NOTCH signaling in many key developmental systems, human mutations in Notch signaling components have been described mainly in disorders with vascular and bone effects. In 2009, Li et al. reported that human pulmonary hypertension could be characterized by the overexpression of NOTCH3 in small PASMCs, and the severity of the disease in humans is correlated with the amount of NOTCH3 protein in the lungs [96]. Recently, Chida et al. identified two novel missense mutations in NOTCH3 in patients with PAH and revealed that these mutations were involved in cell proliferation and viability [97]. The results may contribute to the elucidation of PAH pathogenesis, but whether the NOTCH3 mutations affect NOTCH signaling activity remains unclear. Another report indicated that NOTCH3 mutations in or near the ligand-binding site (EGF-like repeats 10-11) impaired ligand binding sufficiently to affect signaling activity [98]. Furthermore, another two studies suggested that most of the mutations located outside of the ligand-binding site did not impair the signal transduction activity of NOTCH3 [99,100]. Therefore, further investigations are necessary to analyze the function of NOTCH3 in the pathogenesis of PAH.
Adams-Oliver syndrome (AOS) is a rare developmental disorder defined by the combination of aplasia cutis congenita of the scalp vertex and terminal transverse limb defects [101]. In addition, vascular anomalies, such as cutis marmorata telangiectatica congenita, pulmonary hypertension, and retinal hypovascularization, are recurrently observed [102]. Stittrich et al. identified heterozygous variants in NOTCH1 as an additional cause of AOS in multiple families. Phenotypic analysis of affected individuals with NOTCH1 mutations indicates a high rate of vascular anomalies [103]. They found five NOTCH1 mutations in 5 of 12 unrelated families affected by AOS. This condition might lead to loss of function by preventing transcription and translation. Given that NOTCH receptors bind ligands as tetramers [104], it is also possible that some AOS-associated NOTCH1 missense mutations exert dominant-negative effects by destabilizing normal tetramer formation and, thus, disrupt ligand binding. Also conceivable is that some of the NOTCH1 mutations induce a gain of function, which resides in the negative regulatory region, which in turn might reduce the restraint on ligand-independent signaling [105]. The possibility that both loss- and gain-of-function mutations in NOTCH1 cause AOS could be explained if Notch-signaling strength is the critical element in determining normal development of the vasculature [106]. An alternate hypothesis for the different phenotypes observed with AOS-related NOTCH1 mutations is that other genetic, epigenetic, or environmental factors shape the NOTCH1 mutant phenotype.
In summary, there are 13 genes we have known associated with PAH. Nearly half of them involved in TGF-β/BMP signaling pathway including BMPR2, ACVRL1, END, SMAD9 (encodes SMAD 8), SMAD4, and BMP9, which is responsible for the majority of HPAH/IPAH cases. Several other cell signaling pathways contribute to PAH, and their possible resultant signaling or effects of protein actions are briefly listed here (Figure 1).
Genetic testing and counseling
Two consensus guidelines recommend that physicians offer professional counseling on genetics and testing to patients with a history that suggests HPAH [3,107]. Pre-test informed consent and counseling, supported by additional counseling when providing the results, should ensure that all patients involved understand the implications of the results of the testing and what these results might imply for both the patient and family members. Reduced penetrance is one of the many reasons why this is crucial [108]. In addition, the authors of these guidelines have recommended that patients with IPAH be advised about the availability of genetic testing and counseling because of the strong possibility that they carry a disease-causing mutation. The guidelines recommend that professionals offer counseling and testing to the affected IPAH patient before approaching other family members.
Genetic testing for known mutations in PAH-associated autosomal dominant genes is available in North America and Europe for the BMPR2, ACVRL1, ENG, SMAD9, CAV1, and now KCNK3 genes [12], although there currently is no unified PAH mutation panel incorporating all genes. Provision of genetic counseling by trained professionals is vital before and after undertaking clinical genetic testing [109]. Genetic testing is available for either single genes or the entire panel of genes. The cost of testing has recently decreased in the U.S. with the introduction of these gene panels, thereby increasing the accessibility of testing to patients, and usually covered by insurance. Results generally are available in 8 weeks. Genetic testing often is considered in pediatric PAH to explain the etiology of the disease, to counsel family members about identifying other family members at risk and accurately determining the risk of recurrence in future children [14]. Once the mutation in a family is identified, testing other family members for a family-specific mutation in the U.S. is relatively inexpensive, accurate, and fast. Genetic testing is most helpful when it is able to identify members of the family who are not genetically at risk for PAH and who can then forgo the otherwise recommended serial evaluations to screen for the development of PAH.
Affected individuals and "at risk" family members might want to know their mutation status for family planning purposes. Pre-natal screening or pre-implantation diagnosis and management is possible. Reproductive medicine allows several options for preventing transmission of HPAH to the next generation. If the heritable mutation is identified, a medical abortion is an option. Another option is pre-implantation genetic diagnosis and medically-assisted reproduction with selection and implantation of embryos that do not carry the heritable mutation, thereby avoiding the distress of having a medical abortion.
Genetic testing allows identification of pre-symptomatic carriers of PAH-causing mutations who are at high risk of developing PAH. However, it is currently not possible to identify which carriers of a mutation will develop PAH. Thus, genetic testing in relatives will effectively identify mutation noncarriers who have no increased risk of having the heritable disease and potentially provide significant relief; however, mutation carriers currently face many uncertainties. In the U.S., physicians, patients with PAH, and patients' family members have rarely embraced pre-symptomatic genetic testing for several reasons including the unacceptable high cost, discrimination, and the psychological impact of either a positive test (anxiety and depression) or a negative test (survivor guilt). Further studies are needed to analyze the clinical value of noninvasive screening assessments in relatives of patients with IPAH and HPAH and to develop an algorithm for early diagnosis in this cohort [110].
Conclusion
In this review, we discussed all known genes, proteins and their possible mechanisms associated with HPAH or IPAH, which provided a more comprehensive understanding of the complex biological networks and events that promote PAH and provided significant insights into the development of therapeutic interventions for this devastating disorder. However, all the genes identified so far have not fully explained all the HPAH, IPAH, especially the sporadic PAH. We predict that there are more new PAH disease-causing genes or genetic modifiers in the human genome that need to be discovered. Recent advances in the development of NGS for WES or whole genome sequencing (WGS) have provided a powerful tool for the discovery of new disease-causing genes and for the genome-wide association (GWS) studies [111-113]. Despite the fact that the PAH pathogenesis is very complicated, it is promising that large PAH cohort studies in different ethnical populations by the WES or WGS will provide a more complete picture of the underlying genetic architecture for the disease, and may point the way to additional disease mechanisms and therapeutic opportunities.
References
-
Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, et al. (2009) ESC Committee for Practice Guidelines (CPG). Guidelines for the diagnosis and treatment of pulmonary hypertension: the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 30: 2493-2537.
-
Elliott CG (2013) Genetics of pulmonary arterial hypertension. Clin Chest Med 34: 651-663.
-
McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, et al. (2009) ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 119: 2250-2294.
-
Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, et al. (2009) Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 54: S55-66.
-
Barst RJ, Gibbs JS, Ghofrani HA, Hoeper MM, McLaughlin VV, et al. (2009) Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol 54: S78-84.
-
Sitbon O, Humbert M, Jais X, Ioos V, Hamid AM, et al. (2005) Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 111: 3105-3111.
-
Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, et al. (2009) Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 54: S43-54.
-
Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, et al. (2013) Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 62: D34-D41.
-
Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, et al. (2013) Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 62: D42-D50.
-
Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, et al. (2010) Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 122: 156-163.
-
Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg-Maitland M, et al. (2010) Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest 138: 1234-1239.
-
Austin ED, Loyd JE (2014) The genetics of pulmonary arterial hypertension. Circ Res 115: 189-202.
-
Zhang R, Dai LZ, Xie WP, Yu ZX, Wu BX, et al. (2011) Survival of Chinese patients with pulmonary arterial hypertension in the modern treatment era. Chest 140: 301-309.
-
Ma L, Chung WK (2014) The genetic basis of pulmonary arterial hypertension. Hum Genet 133: 471-479.
-
Dresdale DT, Michtom RJ, Schultz M (1954) Recent studies in primary pulmonary hypertension, including pharmacodynamic observations on pulmonary vascular resistance. Bull NY Acad Med 30:195-207.
-
Morse JH, Jones AC, Barst RJ, Hodge SE, Wilhelmsen KC, et al. (1997) Mapping of familial primary pulmonary hypertension locus (PPH1) to chromosome 2q31-q32. Circulation 95: 2603-2606.
-
International PPH Consortium, Lane KB, Machado RD, Pauciulo MW, Thomson JR, et al. (2000) Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet 26: 81-84.
-
Ku CS, Naidoo N, Pawitan Y (2011) Revisiting Mendelian disorders through exome sequencing. Hum Genet 129: 351-370.
-
Piluso G, Aurino S, Cacciottolo M, Del Vecchio Blanco F, Lancioni A, et al. (2010) Mendelian bases of myopathies, cardiomyopathies, and neuromyopathies. Acta Myol 29: 1-20.
-
Dewey FE, Chen R, Cordero SP, Ormond KE, Caleshu C, et al. (2011) Phased whole-genome genetic risk in a family quartet using a major allele reference sequence. PLoS Genet 7: e1002280.
-
Ma L, Roman-Campos D, Austin ED, Eyries M, Sampson KS, et al. (2013) A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 369: 351-361.
-
Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, et al. (2012) Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet 5: 336-343.
-
Germain M, Eyries M, Montani D, Poirier O, Girerd B, et al. (2013) Genome-wide association analysis identifies a susceptibility locus for pulmonary arterial hypertension. Nat Genet 45: 518-521.
-
Miyazono K, Kamiya Y, Morikawa M (2010) Bone morphogenetic protein receptors and signal transduction. J Biochem 147: 35-51.
-
Rabinovitch M (2012) Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 122: 4306-4313.
-
Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, et al. (2000) Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 67: 737-744.
-
International PPH Consortium, Lane KB, Machado RD, Pauciulo MW, Thomson JR, et al. (2000) Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet 26: 81-84.
-
Machado RD, Eickelberg O, Elliott CG, Geraci MW, Hanaoka M, et al. (2009) Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol 54: S32-42.
-
Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, et al. (2000) Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet 37: 741-745.
-
Cogan JD, Pauciulo MW, Batchman AP, Prince MA, Robbins IM, et al. (2006) High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med 174: 590-598.
-
Khajavi M, Inoue K, Lupski JR (2006) Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet 14: 1074-1081.
-
Rosenzweig EB, Morse JH, Knowles JA, Chada KK, Khan AM, et al. (2008) Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J Heart Lung Transplant 27: 668-674.
-
Sztrymf B, Coulet F, Girerd B, Yaici A, Jais X, et al. (2008) Clinical outcomes of pulmonary arterial hypertension in carriers of BMPR2 mutation. Am J Respir Crit Care Med 177: 1377-1383.
-
Grunig E, Weissmann S, Ehlken N, Fijalkowska A, Fischer C, et al. (2009) Stress Doppler Echocardiography in Relatives of Patients With Idiopathic and Familial Pulmonary Arterial Hypertension: Results of a Multicenter European Analysis of Pulmonary Artery Pressure Response to Exercise and Hypoxia. Circulation 119: 1747-1757.
-
Girerd B, Montani D, Eyries M, Yaici A, Sztrymf B, et al. (2010) Absence of influence of gender and BMPR2 mutation type on clinical phenotypes of pulmonary arterial hypertension. Respir Res 11: 73.
-
Austin ED, Phillips JA, Cogan JD, Hamid R, Yu C, et al. (2009) Truncating and missense BMPR2 mutations differentially affect the severity of heritable pulmonary arterial hypertension. Respir Res 10: 87.
-
Wang H, Ji R, Meng J, Cui Q, Zou W, et al. (2014) Functional changes in pulmonary arterial endothelial cells associated with BMPR2 mutations. PLoS One 9: e106703.
-
Greenwald J, Fischer WH, Vale WW, Choe S (1999) Three-finger toxin fold for the extracellular ligand-binding domain of the type II activin receptor serine kinase. Nat Struct Biol 6: 18-22.
-
Machado RD, Southgate L, Eichstaedt CA, et al. (2015) Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum Mutat.
-
Girerd B, Coulet F, Jais X, Eyries M, Van Der Bruggen C, et al. (2015) Characteristics of pulmonary arterial hypertension in affected carriers of a mutation located in the cytoplasmic tail of bone morphogenetic protein receptor type 2. Chest 147: 1385-1394.
-
Watanabe TK, Suzuki M, Omori Y, Hishigaki H, Horie M, et al. (1997) Cloning and characterization of a novel member of the human Mad gene family (MADH6). Genomics 42: 446-451.
-
Massague J, Blain SW, Lo RS (2000) TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 103: 295-309.
-
Nishihara A, Watabe T, Imamura T, Miyazono K (2002) Functional heterogeneity of bone morphogenetic protein receptor-II mutants found in patients with primary pulmonary hypertension. Mol Biol Cell 13: 3055-3063.
-
Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, et al. (2002) Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet 11: 1517-1525.
-
Shintani M, Yagi H, Nakayama T, Saji T, Matsuoka R (2009) A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J Med Genet 46: 331-337.
-
Nasim MT, Ogo T, Ahmed M, Randall R, Chowdhury HM, et al. (2011) Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat 32: 1385-1389.
-
Guttmacher AE, Marchuk DA, White RI Jr (1995) Hereditary hemorrhagic telangiectasia. N Engl J Med 333: 918-924.
-
Shovlin CL, Jackson JE, Bamford KB, Jenkins IH, Kulinskaya E, et al. (2008) Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 63: 259-266.
-
Govani FS, Shovlin CL (2009) Hereditaryhaemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet 17: 860-871.
-
Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, et al. (2001) Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 345: 325-334.
-
Harrison RE, Berger R, Haworth SG, Tulloh R, Mache CJ, et al. (2005) Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation 111: 435-441.
-
Fujiwara M, Yagi H, Matsuoka R, Akimoto K, Furutani M, et al. (2008) Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ J 72: 127-133.
-
Smoot LB, Obler D, McElhinney DB, Boardman K, Wu BL, et al. (2009) Clinical features of pulmonary arterial hypertension in young people with an ALK1 mutation and hereditary hemorrhagic telangiectasia. Arch Dis Child 94:506-511.
-
ten Dijke P, Franzen P, Yamashita H, Ichijo H, Heldin CH, et al. (1994) Serine/threonine kinase receptors. Prog Growth Factor Res 5: 55-72.
-
Girerd B, Montani D, Coulet F, Sztrymf B, Yaici A, et al. (2010) Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med 181: 851-861.
-
Humbert M, Morrell NW, Archer SL, Stenmark KR, Voelkel NF, et al. (2004) Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 43:13S-24S.
-
David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S (2007) Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 109: 1953-1961.
-
tenDijke P, Arthur HM (2007) Extracellular control of TGFbetasignalling in vascular development and disease. Nat Rev Mol Cell Biol 8: 857-869.
-
Rius C, Smith JD, Almendro N, Langa C, Botella LM, et al. (1998) Cloning of the promoter region of human endoglin, the target gene for hereditary hemorrhagic telangiectasia type 1. Blood 92: 4677-4690.
-
Chaouat A, Coulet F, Favre C, Simonneau G, Humbert M, et al. (2004) Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax 59: 446-448.
-
Zeng S, Chen J, Shen H (2010) Controlling of bone morphogenetic protein signaling. Cell Signal 22: 888-893.
-
Fernandez-L A, Sanz-Rodriguez F, Blanco FJ, Bernabeu C, Botella LM (2006) Hereditary hemorrhagic telangiectasia, a vascular dysplasia affecting the TGF-beta signaling pathway. Clin Med Res 4: 66-78.
-
Wooderchak-Donahue WL, McDonald J, O'Fallon B, Upton PD, Li W, et al. (2013) BMP9 Mutations Cause a Vascular-Anomaly Syndrome with Phenotypic Overlap with Hereditary Hemorrhagic Telangiectasia. Am J Hum Genet 93: 530-537.
-
Salani D, Di Castro V, Nicotra MR, Rosano L, Tecce R, et al. (2000) Role of endothelin-1 in neovascularization of ovarian carcinoma. Am J Pathol 157: 1537-1547.
-
Cacoub P, Dorent R, Nataf P, Carayon A, Riquet M, et al. (1997) Endothelin-1 in the lungs of patients with pulmonary hypertension. Cardiovasc Res 33: 196-200.
-
Star GP, Giovinazzo M, Langleben D (2010) Bone morphogenic protein-9 stimulates endothelin-1 release from human pulmonary microvascular endothelial cells A potential mechanism for elevated ET-1 levels in pulmonary arterial hypertension. Microvasc Res 80: 349-354.
-
Park J, Upton PD, De Souza P, Davies RJ, Morrell NW, et al. (2010) Bone Morphogenetic Protein (BMP)-9 Increases Endothelin (ET)-1 Release By Human Pulmonary Artery Endothelial Cells Via A MAPK Dependent Pathway. Am J Respir Crit Care Med 181: A5240.
-
Khoo JP, Zhao L, Alp NJ, Bendall JK, Nicoli T, et al. (2005) Pivotal role for endothelial tetrahydrobiopterin in pulmonary hypertension. Circulation 111: 2126-2133.
-
Lakshminrusimha S, Wiseman D, Black SM, Russell JA, Oishi P et al. (2007) The role of nitric oxide synthase-derived reactive oxygen species in the altered relaxation of pulmonary arteries from lambs with increased pulmonary blood flow. Am J Physiol Heart Circ Physiol 293: H1491-H1497.
-
Achcar RO, Demura Y, Rai PR, Taraseviciene-Stewart L, Kasper M, et al. (2006) Loss of caveolin and heme oxygenase expression in severe pulmonary hypertension. Chest 129: 696-705.
-
Patel HH, Zhang S, Murray F, Suda RY, Head BP, et al. (2007) Increased smooth muscle cell expression of caveolin-1 and caveolae contribute to the pathophysiology of idiopathic pulmonary arterial hypertension. FASEB J 21: 2970-2979.
-
Bauer PM, Yu J, Chen Y, Hickey R, Bernatchez PN, et al. (2005) Endothelial-specific expression of caveolin-1 impairs microvascular permeability and angiogenesis. Proc Natl Acad Sci U S A 102: 204-209.
-
Ju H, Zou R, Venema VJ, Venema RC (1997) Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J Biol Chem 272: 18522-18525.
-
Sowa G, Pypaert M, Sessa WC (2001) Distinction between signaling mechanisms in lipid rafts vs. caveolae. Proc Natl Acad Sci USA 98: 14072-14077.
-
Engelman JA, Zhang XL, Galbiati F, Lisanti MP (1998) Chromosomal localization, genomic organization, and developmental expression of the murine caveolin gene family (Cav-1, -2, and -3). Cav-1 and Cav-2 genes map to a known tumor suppressor locus (6-A2/7q31). FEBS Lett 429: 330-336.
-
Mathew R, Huang J, Shah M, Patel K, Gewitz M, et al. (2004) Disruption of endothelial-cell caveolin-1alpha/raft scaffolding during development of monocrotaline-induced pulmonary hypertension. Circulation 110: 1499-1506.
-
Bauer PM, Bauer EM, Rogers NM, Yao, Feijoo-Cuaresma M, et al. (2012) Activated CD47 promotes pulmonary arterial hypertension through targeting caveolin-1. Cardiovasc Res 93: 682-693.
-
Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, et al. (2012) Whole Exome Sequencing to Identify a Novel Gene (Caveolin-1) Associated with Human Pulmonary Arterial Hypertension Circ Cardiovasc Genet 5: 336-343.
-
de Jesus Perez VA, Yuan K, Lyuksyutova MA, Dewey F, Orcholski ME, et al. (2014) Whole-Exome Sequencing Reveals TopBP1 as a Novel Gene in Idiopathic Pulmonary Arterial Hypertension. Am J Respir Crit Care Med 189:1260-1272.
-
Cescutti R, Negrini S, Kohzaki M, Halazonetis TD (2010) TopBP1 functions with 53BP1 in the G1 DNA damage checkpoint. EMBO J 29: 3723-3732.
-
Reyes R, Duprat F, Lesage F, Fink M, Salinas M, et al. (1998) Cloning and expression of a novel pH-sensitive two pore domain K+ channel from human kidney. J Biol Chem 273: 30863-30869.
-
D'Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, et al. (1991) Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 115: 343-349.
-
Gardener MJ, Johnson IT, Burnham MP, Edwards G, Heagerty AM, et al. (2004) Functional evidence of a role for two-pore domain potassium channels in rat mesenteric and pulmonary arteries. Br J Pharmacol 142: 192-202.
-
Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, et al. (2007) Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol 292: C1837-1853.
-
Morecroft I, Murray A, Nilsen M, Gurney AM, MacLean MR (2009) Treatment with the Kv7 potassium channel activator flupirtine is beneficial in two independent mouse models of pulmonary hypertension. Br J Pharmacol 157: 1241-1249.
-
Wang G, Knight L, Ji R, Lawrence P, Kanaan U, et al. (2014) Early onset severe pulmonary arterial hypertension with 'two-hit' digenic mutations in both BMPR2 and KCNA5 genes. Int J Cardiol 177: e167-169.
-
Mandel J, Mark EJ, Hales CA (2000) Pulmonary veno-occlusive disease. Am J Respir Crit Care Med 162: 1964-1973.
-
Montani D, Price LC, Dorfmuller P, Achouh L, Jais X, et al. (2009) Pulmonary veno-occlusive disease. Eur Respir J 33: 189-200.
-
Eyries M, Montani D, Girerd B, Perret C, Leroy A, et al. (2014) EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nature Genetics 46: 65-69.
-
Donnelly N, Gorman AM, Gupta S, Samali A (2013) The eIF2a kinases: their structures and functions. Cell Mol Life Sci 70: 3493-3511.
-
Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, et al. (2001) BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet 68: 92-102.
-
Barrios-Rodiles M, Brown KR, Ozdamar B, Bose R, Liu Z, et al. (2005) High-throughput mapping of a dynamic signaling network in mammalian cells. Science 307: 1621-1625.
-
Artavanis-Tsakonas S, Rand MD, Lake RJ (1999) Notch signaling: cell fate control and signal integration in development. Science 284: 770-776.
-
Wang T, Baron M, Trump D (2008) An overview of Notch3 function in vascular smooth muscle cells. Prog Biophys Mol Biol 96: 499-509.
-
Li X, Zhang X, Leathers R, Makino A, Huang C, et al. (2009) Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med 15: 1289-1297.
-
Chida A, Shintani M, Matsushita Y, Sato H, Eitoku T, et al. (2014) Mutations of NOTCH3 in childhood pulmonary arterial hypertension. Mol Genet Genomic Med 2: 229-239.
-
Joutel A, Monet M, Domenga V, Riant F, Tournier-Lasserve E (2004) Pathogenic mutations associated with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy differently affect Jagged1 binding and Notch3 activity via the RBP/JK signaling pathway. Am J Hum Genet 74: 338-347.
-
Peters N, Opherk C, Zacherle S, Capell A, Gempel P, et al. (2004) CADASIL-associated Notch3 mutations have differential effects both on ligand binding and ligand-induced Notch3 receptor signaling through RBP-Jk. Exp Cell Res 299: 454-464.
-
Low WC, Santa Y, Takahashi K, Tabira T, Kalaria RN (2006) CADASIL-causing mutations do not alter Notch3 receptor processing and activation. Neuroreport 17: 945-949.
-
Monet-Lepretre M, Bardot B, Lemaire B, Domenga V, Godin O, et al. (2009) Distinct phenotypic and functional features of CADASIL mutations in the Notch3 ligand binding domain. Brain 132: 1601-1612.
-
Martinez-Frias ML, Arroyo Carrera I, Munoz-Delgado NJ, Nieto Conde C, Rodriguez-Pinilla E, et al. (1996) The Adams-Oliver syndrome in Spain: the epidemiological aspects. An Esp Pediatr 45: 57-61.
-
Snape KM, Ruddy D, Zenker M, Wuyts W, Whiteford M, et al. (2009) The spectra of clinical phenotypes in aplasia cutis congenita and terminal transverse limb defects. Am J Med Genet A 149A: 1860-1881.
-
Stittrich AB, Lehman A, Bodian DL, Ashworth J, Zong Z, et al. (2014) Mutations in NOTCH1 cause Adams-Oliver syndrome. Am J Hum Genet 95: 275-284.
-
Hambleton S, Valeyev NV, Muranyi A, Knott V, Werner JM, et al. (2004) Structural and functional properties of the human notch-1 ligand binding region. Structure 12: 2173-2183.
-
Tsuji H, Ishii-Ohba H, Ukai H, Katsube T, Ogiu T (2003) Radiation-induced deletions in the 5' end region of Notch1 lead to the formation of truncated proteins and are involved in the development of mouse thymic lymphomas. Carcinogenesis 24: 1257-1268.
-
Petrovic J, Formosa-Jordan P, Luna-Escalante JC, Abello G, Ibanes M, et al. (2014) Ligand dependent Notch signaling strength orchestrates lateral induction and lateral inhibition in the developing inner ear. Development 141: 2313-2324.
-
Badesch DB, Abman SH, Simonneau G, Rubin LJ, McLaughlin VV (2007) Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest 131:1917-1928.
-
Larkin EK, Newman JH, Austin ED, Hemnes AR, Wheeler L, et al. (2012) Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 892-896.
-
Newman JH, Trembath RC, Morse JA, Grunig E, Loyd JE, et al. (2004) Genetic basis of pulmonary arterial hypertension: current understanding and future directions. J Am Coll Cardiol 43: 33S-39S.
-
Soubrier F, Chung WK, Machado R, Grunig E, Aldred M,et al. (2013) Genetics and Genomics of Pulmonary Arterial Hypertension. J Am Coll Cardiol 62: D13-21.
-
Chan SY, Loscalzo J (2012) The emerging paradigm of network medicine in the study of human disease. Circ Res 111: 359-374.
-
Liu D, Morrell NW (2013) Genetics and the molecular pathogenesis of pulmonary arterial hypertension. Curr Hypertens Rep 15: 632-637.
-
Wu DC, Zhang HD, Jing ZC (2013) Pediatric pulmonary arterial hypertension. Curr Hypertens Rep 15: 606-613.
