

International Journal of Respiratory and Pulmonary Medicine
Debilitating Disease in a Daring Child with Determined Parents - Impact on Survival
Marie Christy Sharafine Stephen1*, Eswaran Venkata Raman1, Gnanam Aram2 and Anitha Kumari1
1Department of Otolaryngology Head & Neck Surgery, Manipal Hospitals, India
2IAP/ISCCM Fellowship in Pediatric Critical Care, Manipal Hospitals, India
*Corresponding author: Dr. Marie Christy Sharafine Stephen, Department of Otolaryngology Head & Neck Surgery, Manipal Hospitals, No: E906, Alpine Eco Apartments, Doddenakundi, Marathahalli, Bengaluru - 560037, India, Tel: 8095128110, E-mail: sharafine@gmail.com
Int J Respir Pulm Med, IJRPM-3-037, (Volume 3, Issue 1), Case Report; ISSN: 2378-3516
Received: January 05, 2016 | Accepted: January 30, 2016 | Published: February 02, 2016
Citation: Stephen MCS, Raman EV, Aram G, Kumari A. (2016) Debilitating Disease in a Daring Child with Determined Parents - Impact on Survival. Int J Respir Pulm Med 3:037. 10.23937/2378-3516/1410037
Copyright: © 2016 Stephen MCS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution,
and reproduction in any medium, provided the original author and source are credited.
Abstract
Pompe disease is a rare and progressive lysosomal storage disorder, of autosomal recessive inheritance. Disease onset and progression range from early infantile onset which is fatal and rapidly progressive to the late onset formthatis slowly progressive, but leads to severe respiratory dysfunction and significant morbidity. Besides, scoliosis and other spine deformities cause substantial workload on the already compromised ventilation. This has a major impact on the quality of life and life-expectancy. Enzyme replacement therapy has in recent times, promised longer survival. We report a teenager with Pompe disease with severe respiratory compromise and spine deformity, who has survived the disease on enzyme replacement therapy and tracheostomy with home positive pressure ventilation (BiPAP) for more than a decade now. We report this case in view of its rarity, the reasonablyfavorable clinical course and better quality of life in this patient.
Keywords
Pompe, Storage disorder, Scoliosis, Tracheostomy, Enzyme replacement
Abbreviations
BiPAP: Bilevel Positive Airway Pressure; PCR: Polymerase Chain Reaction
Introduction
Respiratory failure is a major cause of morbidity and mortality in many of the neuromuscular disorders. Pompe disease is one such glycogen storage disorder which leads to frequent respiratory compromise, either at the disease onset or during advanced stages and may progress more rapidly than the motor impairment [1]. 70% of patients develop progressive respiratory dysfunction with a mean reduction in vital capacity (VC) ranging from 0.9-4.5% [1]. The likelihood of requiring mechanical ventilation increases by an average of 8% every year following diagnosis [1]. Mechanical ventilation is often required during an episode of acute respiratory failure without any prior diagnosis of respiratory problems. We report a patient who was diagnosed with Pompe disease at 7 years of age while being evaluated for failed extubation for pneumonia. She required tracheostomy and BiPAP, later developed spine deformity, but leads a better quality of life for the disease process.
Case Report
7 year old girl presented to the emergency department with acute onset fever and respiratory distress. She was the first child of a non-consanguineous couple, born by LSCS at term with no perinatal complications. She had attained her initial milestones on time except her motor milestones which were delayed. She was diagnosed to have pneumonia and in view of impending respiratory failure, she was intubated and required mechanical ventilation. Following resolution of pneumonia, there was difficulty in weaning her from the ventilator.
She was suspected to have some neuromuscular disorder and in view of prolonged ventilation anticipated, she underwent Tracheostomy (size 5 Portex tube, uncuffed) and was maintained on BiPAP respiratory support. She tolerated the same well. Further investigation in search of diagnosis was undertaken. Biochemical urinalysis revealed elevated hexose tetrasaccharide fraction (urinary Hex4) - 98 mmol/mol creatinine (normal: < 5). While this may be associated with glycogen storage disorders (III, VI and II) and muscular dystrophies, confirmation was awaited via gene sequencing. Her blood sample at the Molecular Diagnostics Lab [(Department of pathology), Duke University Health system, Durham] was sequenced by PCR for mutations in the acid alpha glucosidase (GAA) genethat encodes the enzyme acid alpha glucosidase. She was found to be heterozygous for 14 known single base pair polymorphisms and for two previously reported mutations in the GAA gene consistent with Pompe disease with deficient enzyme activity:a heterozygous missense mutation in exon 3 (c. 670C > T), that alters the arginine codon at position 224 to a tryptophan codon which results in decreased enzyme activity (acid alpha glucosidase) and aheterozygous nonsense mutation in exon 18 (c. 2608C > T), that alters the arginine codon position 870 to a stop codon which results in a premature termination codon [2,3].
In view of this, she received enzyme replacement therapy with Myozymeinfusions (slow IV infusions of prepared solution; each vial 50 mg) at 20 mg/kg every 15 days, as per published guidelines. Though she initially had some infusion related minor reactions, she gradually tolerated it well. Her IgG antibodies to Myozyme were periodically monitored.
She was noted to have regression of motor milestones after her 7th birthday and developed difficulty in walking, though her remaining milestones were preserved. 2 years later, she noticed pain with gradual deformity of her backand with progressive muscle weakness, she became wheel chair bound. She was found to have developed neuromuscular thoracolumbar kyphoscoliosis with fixed pelvic obliquity and sitting imbalance. She underwent Spino-pelvic instrumented scoliosis correction and fusion T1-ilium under general anesthesia. Following this spine stabilization surgery, she was continued on Tracheostomy with BiPAP support at home and enzyme infusions. Her feeding and nutrition were normal. Her echocardiogram showed stable cardiac function. Bronchoscopy showed normal vocal cords with tracheomalacia above the stoma level (Figure 1).
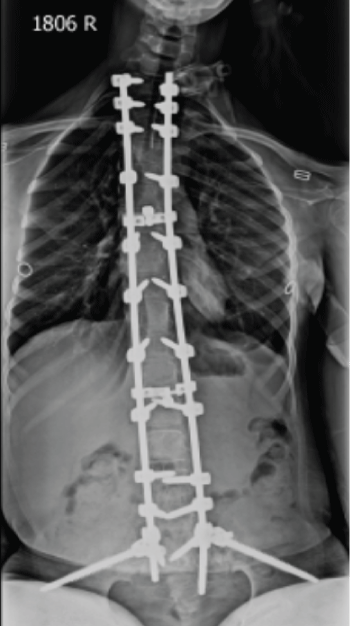
.
Figure 1: X-ray chest and spine: post-op spine stabilization surgery for kyphoscoliosis and fixed pelvic deformity.
View Figure 1
Her parents who were the caregivers were explained about the progressive nature of the disease and the respiratory embarrassment associated with the quality of life. They were well motivated, were gradually trained on tracheostomy care, its maintenance and were educated on BiPAP andits appropriate change of settings when required. She received bimonthly enzyme infusions (Genzyme) and periodic tracheostomy tube changes. She was managed effectively at home with tracheal tube suctioning as required and stoma care. Except for her bimonthly visits to hospital for enzyme infusion, she has not had frequent respiratory tract infections or tracheostomy tube related complications (Figure 2).
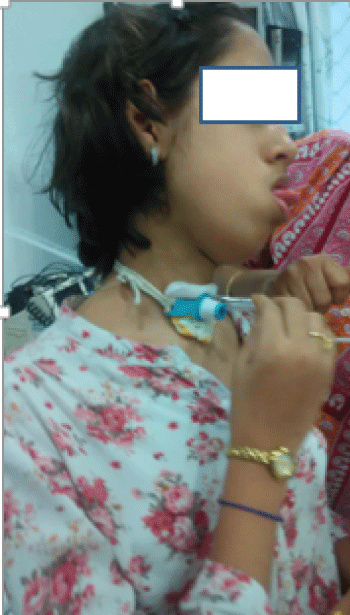
.
Figure 2: Clinical picture of patient doing self-suctioning of the tracheostomy tube.
View Figure 2
The parents, not only provided adequate care for her, but also trained her self-care and be self-dependent, if not for the motor disability. Though she was wheel-chair bound, she managed her daily activities and was active in educational accomplishments. She had completed her schooling and is currently a 16 year old teenager who is pursuing further studies at home, at the time of this write-up. She has managed the disease effectively well despite being restricted to the wheel chair (Figure 3).
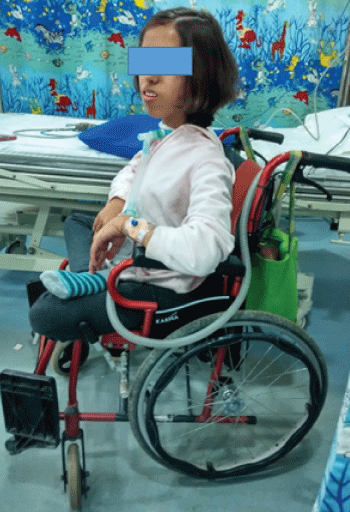
The disease had changed, not just the patient’s quality of life, but that of her parents, such that they have initiated a private non-governmental organization, dedicated to the care of children with such rare diseases.
Discussion
Pompe disease is a rare and progressive autosomal recessive disorder. It is also called as acid maltase deficiency or glycogen storage disorder type II. It is caused by a genetic mutation in the acid alpha glucosidase gene leading to a deficient enzyme, acid alpha-glucosidase (GAA) [4]. This results in progressive accumulation of lysosomal glycogenin multiple tissues, cardio-respiratory and skeletal muscle being the most severely affected. The disease shows onset at different ages and with different degrees of severity, usually correlate well with the nature of the mutation and the degree of residual enzymatic activity [5]. The incidence ranges from 1 in 33,000 to 1 in 300,000 [1].
Disease onset and progression can range three types: the classic early-onset, non-classic infantile onset and late-onset (childhood, juvenile and adult). The early onset classic infantile disease may be apparent in-utero, but often present in the first two months of age with generalized muscle weakness, hypotonia, cardiomegaly with hypertrophic cardiomyopathy, feeding difficulties, failure to thrive, respiratory distress and hearing loss. Without enzyme replacement therapy, it frequently culminates in death by the child’s first birthday from progressive left ventricular outflow obstruction. The infantile onset, non-classic variant manifest by the first year of life with motor delays, slowly progressive muscle weakness and classically result in death due torespiratory failure in early childhood. Cardiomegaly though occur, is not a major source of morbidity [6]. Late-onset disease can manifest as early as first or as late as sixth decade, typified by progressive proximal muscle weakness and respiratory failure without cardiac involvement.
Management guidelines are established. Individualized care for cardiomyopathy and appropriate physiotherapy to maintain range of motion and promote ambulation, surgery for contracture if needed, besides nutrition and feeding support [6].
Respiratory dysfunction develops in ~ 70% of patients with respiratory failure being the single most cause of significant morbidity and mortality. Progressive respiratory muscle weakness develop in all three groups of muscles: inspiratory, expiratory and bulbar muscles. In view of this, there is impaired ventilation, decreased clearance of airway secretions and cough, besides risk of aspiration. This also exposes them to frequent respiratory infections that further debilitates the already compromised respiratory system. As a result of this, the quality of life of both the patient and the care givers is highly impaired. Respiratory support in these patients may require inspiratory/expiratory muscles, CPAP, BiPAP and/or tracheostomy.
Of late, patients with Pompe disease are reported to frequently develop spine deformities. This could be related to the longer survival ensured by the available enzyme substitution therapy. 6 out of 8 patients with Pompe disease, in a case series revealed orthopedic problems of scoliosis, hip dysplasia, feet deformities, and contractures, all requiring operative intervention [7]. Another study showed presence of scoliosis in one third of all patients and in them the disease onset was seen as children (≥ 2 to < 13 years) and juveniles (≥ 13 to < 20 years). Patients with scoliosis were diagnosed with Pompe disease for a mean of 1.2 years before the first reporting of scoliosis and 62% was reported in wheel chair bound patients [8]. It has been observed that there is a relative resistance of skeletal muscle to effective glycogen depletion with enzyme infused that varies with different enzyme preparations. In addition, high IgG titers to alpha glucosidase, increase in muscle glycogen therapy and CRIM (cross-reactive immunologic material) negativity are some of the factors that have been associated with progressive disease and poor prognosis [6].
Enzyme replacement therapy with Myozyme (2006 FDA approved) or Lumizyme (2010 FDA approved) which contains alglucosidase alfa is commercially available and represents the only available etiologic therapy for this disease [6]. This substitutive enzyme is industrially manufactured through biotechnological process and administered through intravenous infusion. It is also effective in preventing Pompe disease progression in late-onset patients. Thus it is recommended to begin therapy as soon as the diagnosis is established [4].
Our patient was diagnosed at the age of 7 during an episode of lower respiratory tract infection. Besides progressive neuromuscular weakness, she also developed fixed pelvic and spine deformities with kyphoscoliosis which added further to the respiratory embarrassment. She was found to have high IgG titers of antibody to Myozyme which could have added to her worsening spine deformity (resistance of skeletal muscle to enzyme infusion) although progressive muscle weakness associated with the disease per-se cannot be undermentioned. Following surgical correction, she continued on her enzyme infusions and showed a stable ventilator function and motor ability. She also had reduced frequency of respiratory infections, besides better nutrition and feeding. Above all, her quality of life was reasonably favorable for the nature of this devastating disease. Also, it had created a great deal of impact on her parents’ lifestyle that they involved themselves on a full-time charitable organization dedicated to the care of such children.
Conclusion
Pompe disease is a debilitating progressive neuromuscular disorder with variable clinical presentation. The late onset disease, though slowly progressive leads to significant morbidity especially economical and emotional burden to caregivers. Life expectancy is recently being prolonged with substitutive enzyme therapy and the quality of life can be improved by patient education and motivation besides dedicated caregivers.
References
-
Nicolino Ambrosino, Marco Confalonieri, Grazia Crescimanno, Andrea Vianello, Michele Vitacca (2013) The role of respiratory management of Pompe disease. Respiratory Medicine 107: 1124-1132.
-
Pittis MG, Montalvo AL, Miocic S, Martini C, Deganuto M, et al. (2003) Identification of four novel mutations in the alpha glucosidase gene in five Italian patients with infantile onset glycogen storage disease type II.Am J Med Genet A 121A: 225-230.
-
McCready ME, Carson NL, Chakraborty P, Clarke JT, Callahan JW, et al. (2007) Development of a clinical assay for detection of GAA mutations and characterization of the GAA mutation spectrum in a Canadian cohort of individuals with glycogen storage disease, type II.Mol Genet Metab 92: 325-335.
-
Ley-Martos M, Salado-Reyes MJ, Espinosa-Rosso R, Solera-Garcia J, Jimenez-Jimenez L (2015)Variability in the clinical presentation of Pompe disease: development following enzyme replacement therapy. Rev Neurol Nov 61: 416-420.
-
Lim J-A, Li L, Raben N (2014) Pompe disease: from pathophysiology to therapy and back again. Front Aging Neurosci.
-
Leslie N, Tinkle BT (2016) Glycogen Storage Disease Type II (Pompe Disease) GeneReviews(®) [Internet].
-
Haaker G, Forst J, Forst R, Fujak A (2014) Orthopedic management of patients with Pompe disease: a retrospective case series of 8 patients. ScientificWorld Journal.
-
Roberts M, Kishnani PS, van der Ploeg AT, Müller-Felber W, Merlini L et al. (2011) The prevalence and impact of scoliosis in Pompe disease: lessons learned from the Pompe Registry. Mol Genet Metab 104: 574-582.
