

Journal of Genetics and Genome Research
Aspergillus flavus Blast2GO Gene Ontology Database: Elevated Growth Temperature Alters Amino Acid Metabolism
Perng-Kuang Chang* and Leslie L. Scharfenstein
Southern Regional Research Center, Agricultural Research Service, U. S. Department of Agriculture, USA
*Corresponding author:
Perng-Kuang Chang, Southern Regional Research Center, Agricultural Research Service, U. S. Department of Agriculture, 1100 Robert E. Lee Boulevard, New Orleans, Louisiana 70124, USA, Tel: 504-286-4208; Fax: 504-286-4419; E-mail: perngkuang.chang@ars.usda.gov
J Genet Genome Res, JGGR-1-005, (Volume 1, Issue 1), Research Article; ISSN: 2378-3648
Received: September 04, 2014 | Accepted: October 01, 2014 | Published: October 05, 2014
Citation: Chang P-K, Scharfenstein LL (2014) Aspergillus flavus Blast2GO Gene Ontology Database: Elevated Growth Temperature Alters Amino Acid Metabolism. J Genet Genome Res 1:005. 10.23937/2378-3648/1410005
Copyright: © 2014 Chang P-K, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The availability of a representative Gene Ontology (GO) database is a prerequisite for a successful functional genomics study. Using the online Blast2GO tool we constructed a GO database of Aspergillus flavus, a plant and human pathogen. Of the predicted total 13,485 A. flavus genes 8,987 were annotated with GO terms. The mean GO level was 5.64. Using a low stringency setting of a sequence cut-off number of 10 and a node score of 20, we obtained 1,177 GO terms associated with biological process, 388 GO terms associated with molecular function and 200 GO terms associated with cellular component. Of the 8,987 annotated genes 4,232 were mapped to 129 reference pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The utility of the GO database was evaluated with published transcriptomic data by assessing physiological states in relation to the metabolic capacity of A. flavus growing at 30°C and 37°C. Results showed that growth at 30°C favored degradation of amino acids with nonpolar side chains, such as valine, leucine, isoleucine, phenylalanine and tryptophan. In contrast, growth at 37°C favored the degradation of arginine and histidine (basic), aspartate and glutamate (acidic), and serine and threonine (uncharged but polar) but biosynthesis of the aforementioned nonpolar-side-chain amino acids. KEGG pathways of amino acid degradation contributing to acetyl-CoA production, (saturated) fatty acid biosynthesis and degradation as well as biosynthesis of unsaturated fatty acids were active at 30°C, which suggests a requirement that A. flavus maintain a high content of unsaturated fatty acids at the optimal growth temperature. The proposition was supported by the finding that the GO cellular component involved at 30°C growth was mainly a fatty acid synthase complex. The constructed A. flavus GO database was proven to be useful in our functional genomics study. We also outlined the procedures for future update and refinement of the current A. flavus GO database, which can be achieved by concerted efforts of the Aspergillus research community.
Keywords
Aspergillus, Gene ontology, Transcriptome, Blast2GO, KEGG, Temperature, Amino acid metabolism
Introduction
Aspergillus flavus is predominately a saprophytic fungus in the soil that grows on dead plant, agricultural debris, and animal tissue. It can infect many crops such as corn, cotton, peanuts, and nut trees and the infection often leads to contamination with aflatoxin B1, a toxic and potent carcinogenic compound. Because of its ability to grow at 37°C, it also is an opportunistic pathogen for humans and animals and is known to be the second leading cause of invasive aspergillosis in humans [1]. The genome of A. flavus NRRL3357, which consists of eight chromosomes, has been sequenced and its size determined to be about 37 Mb [2]. The genome sequence of another A. flavus strain AF70S (ATCC MYA-384), which produces small sclerotia, has also been annotated and assembled but the data are not yet available to the public (Yu, personal communication). Seven additional A. flavus genome sequences not yet assembled also are available [3]. Comparative genome studies have been performed to decipher evolutionary relationship among A. flavus and related species [2-4]. Genome-wide investigations using EST or whole-genome microarray assays have been performed to examine genes differentially expressed in degenerated A. flavus strains after serial transfers [5], in a 70S-derived strain that is defective in the veA regulatory gene [6], in NRRL3357 that examined tryptophan supplementation on aflatoxin biosynthesis [7], in the simulated conditions of A. flavus-host interactions [8] and in the process of A. flavus-seed colonization [9]. However, interpretations of the research results, more often than not, have not provided meaningful conclusions to the studied conditions. The inability to pertinently interpret the results is mainly due to the lack of knowledge in network interactions among the differentially expressed genes examined.
With the advent of the next generation sequencing of RNA (RNA-Seq) [10], functional genomics research in many organisms has expanded enormously in recent years. The application of RNA-Seq routinely generates unprecedented amounts of data on gene expression. To understand the transcriptional landscape and associated biological meanings, researchers have embraced bioinformatic technologies that categorize genes that are differentially expressed under studied conditions. One technique widely used to highlight the biological implications is to apply Gene Ontology (GO) enrichment analysis. This approach groups differentially expressed genes into categories based on predefined biological properties. It then statistically tests each category against the non-differentially expressed gene population to determine whether a category is over represented among the differentially expressed genes. GO is a collection of defined terms that represent gene product properties. It covers three main domains: 1) biological process, which represents operations or molecular events with a defined beginning and end; 2) molecular function, which describes gene product activities at the molecular level; and 3) cellular component, which indicates parts of a cell or its extracellular environment (http://www.geneontology.org). Many web-based tools are available for performing GO analysis, for example, Blast2GO [11], DAVID [12], ClueGO [13], AmiGO (http://www.geneontology.org), AgriGO [14], and FungiFun [15]. Among these, FungiFun is designed specifically for functional characterization of fungal genes, and Blast2GO has great versatility with statistically robust tests that aim to eliminate false-positive hits.
Although transcriptomic profiling by RNA-Seq has been carried out in A. flavus [16,17], functional genomics analyses have been lacking due to the unavailability of a suitable A. flavus GO database. To establish a GO database for A. flavus that can be freely shared by research groups in the form of an annotation file, we analyzed all predicted 13,485 genes of A. flavus NRRL3357 and obtained GO terms of the annotated genes using the free online resource of BLAST2GO (http://www.blast2go.com). The annotation file can be requested from the corresponding author. The utility of the database was evaluated with available RNA-Seq data previously used to investigate temperature effects on aflatoxin biosynthesis of A. flavus [17]. We used these data to further assess the metabolic states of A. flavus growing at 30°C and 37°C. The results showed that different groups of amino acids were utilized at different growth temperatures. The higher pathway activities of amino acid metabolism that produces acetyl-CoA, (saturated) fatty acid biosynthesis and degradation, and biosynthesis of unsaturated fatty acids at 30°C than at 37°C may account for the high content of unsaturated fatty acids in A. flavus, which comprises 70 to 75% of the total fatty acids [18]. The constructed A. flavus GO database although proven to be useful is still far from perfect. Procedures for future update and refinement of the current database for use in A. flavus and closely related aspergilli were outlined.
Materials and Methods
Construction of the A. flavus GO database
The A. flavus NRRL3357 genome sequence and annotations were acquired from NCBI (http://www.ncbi.nlm.nih.gov). The online resource Blast2GO (http://www.blast2go.com/b2ghome) was used to assign GO terms to A. flavus NRRL3357 gene products [11,19]. All 13,485 genes of A. flavus NRRL3357 was analyzed by a BlastX search against the NCBI non-redundant (nr) database with an Expect (E) value =1.0E-3 and a maximum of 20 hits for each gene. In the mapping step default weights of the evidence codes were used. In the annotation step only the gene hits with an E value =1.0E-6 were further analyzed. An annotation score of 55 was used as the cutoff value after a GO-Weight of 5 was given to mapped children terms. An InterProScan search for conserved domains and motifs was performed next followed by the use of the Annex function to augment the GO terms.
GO analysis and KEGG pathway assignmentsThe RNA-Seq data from the study of temperature effects on genome-wide gene expression of A. flavus NRRL3357 [17] were obtained from the NCBI's GEO database, which is under the accession number, GSE30031. Functional GO enrichment analyses were performed on annotations of up-regulated (= four-fold) genes at 30°C or 37°C using Fisher's Exact Test with Multiple Test Correction of False Discovery Rate (FDR) at the significance threshold of 0.05. The setting of FDR < 0.05 is in general 1,000 times more stringent than the p-valve < 0.05 [20]. Both (i) the differentially expressed genes, that is, those up-regulated =four-folded at 30°C or 37°C, and (ii) the GO-enriched and annotated genes with FDR < 0.05 at 30°C or 37°C were analyzed using the reference metabolic pathways of the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [21].
Results
GO annotation of A. flavus genes
We first performed a BlastX search against the NCBI non-redundant (nr) database with an Expect (E) value =1.0E-3 and a 20 hit maximum. Of the 13,485 predicted genes 12,742 (94.5%) met the selection criteria. Those genes that did not meet the selection criteria included 141 with no homology (HSP length cutoff=33) to proteins in the nr database. The majority of the hits had E values =1.0E-25 with degrees of homology ranging from 65% to 100%. Of the 12,742 selected genes 12,194 (95.7%) were mapped sequences. GO annotation, which applies a specific annotation rule on obtained ontology terms, resulted in 31,194 annotations and a mean GO level of 5.66 from a pool of 8,519 annotated sequences. In this step, the number of sequences with an enzyme code (EC) was 2,952 and 3,712 ECs were obtained. The next InterProScan step, which retrieves functional domains or motifs from amino acid sequences, increased the total annotations to 32,714. Among these 28,206 were confirmed annotations, and 4,508 were too general and were removed. The further Annex step, which increases the annotation density but does not increase the number of annotated sequences, enriched the annotations by 4,445 (13.6%). The overall analysis resulted in 8,987 A. flavus sequences annotated with 37,139 GO terms. Figure 1 shows the distribution of annotation, i.e., number of GO term in relation to the total number of the A. flavus sequences, annotation score, and GO level.
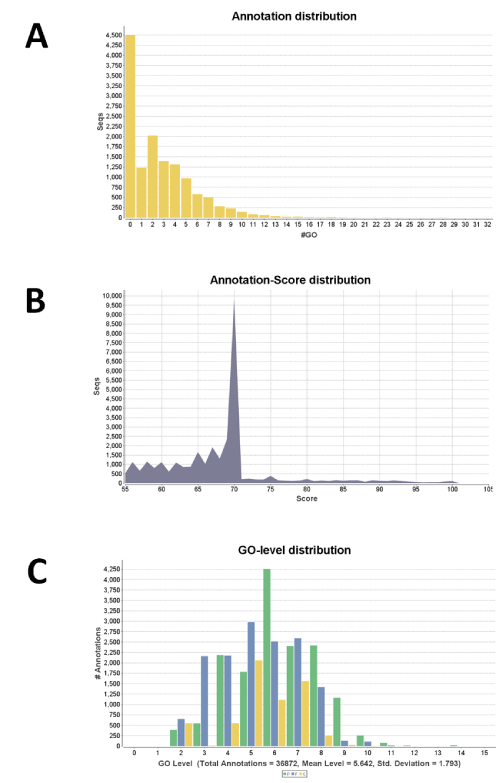 (A) GO term number in relation to the total number of the A. flavus
sequences; (B) Annotation score in relation to the number of sequences;
and (C) GO level in relation to the number of annotation.
(A) GO term number in relation to the total number of the A. flavus
sequences; (B) Annotation score in relation to the number of sequences;
and (C) GO level in relation to the number of annotation.
Figure 1: Summary of the distribution of annotations of A. flavus.
View Figure 1
GO functional categories and KEGG metabolic pathways of A. flavus
The graphical presentation of the three main GO functional categories: Biological process, molecular function and cellular component allows visualization of grouped annotated genes to reveal associated biological functions. Parameters in Blast2GO such as Sequence Number Filter and Node Score Filter can be set to control the graphic content, which in turn affects the numbers of GO terms tabulated for the three functional categories. Using a preliminary setting of a sequence number of 10 and a node score of 20, we obtained 1,177 GO terms associated with biological process, 388 GO terms associated with molecular function and 200 GO terms associated with cellular component. Biological process, molecular function, and cellular component are each the ultimate parent of its own functional category and are treated as "level one" GO terms. When the GO level, sequence number and node score were arbitrarily set to intermediate ranges (Figure 2), the number of GO terms in each functional category was substantially decreased. Under these specific criteria, organic substance metabolic process (GO:0071704), cellular metabolic process (GO:0044237), and primary metabolic process (GO:0044238) are the main categories of the biological process, each with over 4,000 genes. Organic cyclic compound binding (GO:0097159), heterocyclic compound binding (GO:1901363), and ion binding (GO:0043167) are the major binding activities of the molecular function, each with over 2,500 genes. For the cellular component, intracellular part (GO:0044424), cell part (GO:0044464), and intracellular (GO:0005622) form the main categories, each also with over 2,500 genes. To identify metabolic pathways of A. flavus, we mapped the 8,987 annotated genes to the reference pathways in the KEGG database. Of the annotated genes, 4,232 were assigned to 129 pathways. The pathways with over 100 genes annotated included those for metabolism of purine (222), starch and sucrose (138), and amino acids of glycine, serine and threonine (116) (Supplemental Material).
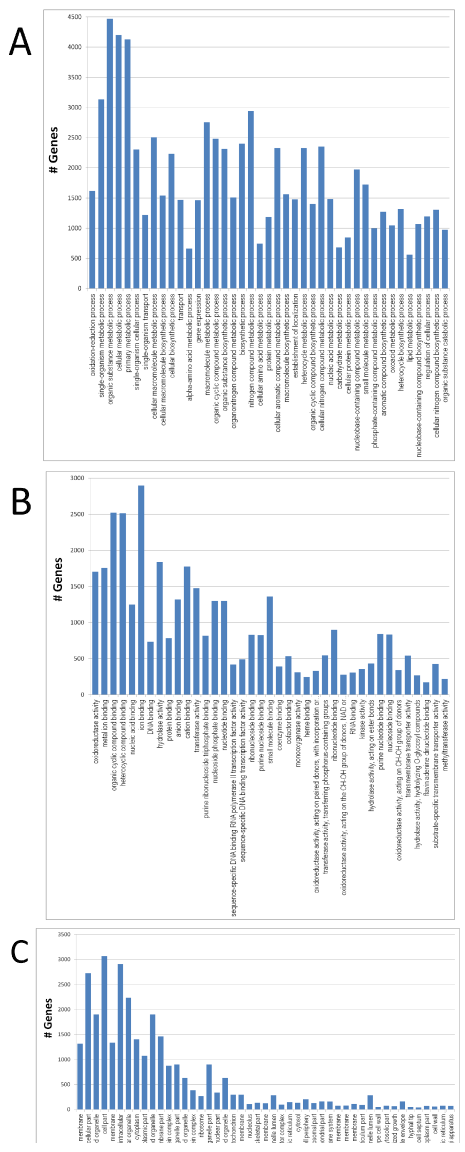 (A) Biological process: GO level was set at three to five, sequence cut-off
number was 450 and node score cut-off was 200; (B) Molecular function:
GO level was set at three to five, sequence cut-off number was 150 and
node score cut-off was 150; and (C) Cellular component: GO level was
set at three to six, sequence cut-off number was 15 and node score
cut-off was 35.
(A) Biological process: GO level was set at three to five, sequence cut-off
number was 450 and node score cut-off was 200; (B) Molecular function:
GO level was set at three to five, sequence cut-off number was 150 and
node score cut-off was 150; and (C) Cellular component: GO level was
set at three to six, sequence cut-off number was 15 and node score
cut-off was 35.
Figure 2: GO functional categories of biological process, molecular
function, and cellular component for A. flavus.
View Figure 2
Effects of growth temperatures on A. flavus GO functional categories and KEGG pathways
The utility of the constructed A. flavus GO database was evaluated with RNA-Seq data that examined temperature effects on expression of genes of the aflatoxin gene cluster [17]. Using the dataset we calculated that 12,081 (89.6%) genes were expressed for all samples combined and 10,594 (78.6%) genes were expressed at both 30°C and 37°C. In this study, using fold-of-increase =four as the criterion, we found that a total of 1,973 genes were differentially expressed under these conditions with 1,259 genes up-regulated at 30°C and 714 genes up-regulated at 37°C. Among these 849 of the 1,259 genes and 473 of the 714 genes were annotated with GO terms. GO enrichment analyses indicated that many biological processes and molecular functions were negatively affected at 37°C. In other words, the activities of the corresponding processes and functions at 30°C were relatively enhanced. Figure 3 shows that catabolism for specific amino acids, such as valine, leucine, isoleucine, tyrosine, phenylalanine, alanine, and tryptophan, fatty acid biosynthesis, metabolism of simple carbon compounds such as acetate and propionate, and electron transport were elevated at 30°C. The cellular component involved was the fatty acid synthase complex. KEGG metabolic pathway analysis concluded that the annotated genes up-regulated at 30°C were associated with 104 pathways. At 37°C only the molecular function of transferase activity was enriched (data not shown). Nonetheless, 81 KEGG pathways were associated with the up-regulated annotated genes. To investigate how growth temperatures affected A. flavus metabolism, we arbitrarily compared the top 10 metabolic pathways associated with growth at 30°C and those with growth at 37°C, which contained approximately 5% and 1% of the annotated sequences, respectively (Table 1). We further analyzed KEGG pathways associated with GO-enriched, annotated sequences for growth at 30°C and at 37°C. The top 10 pathways enriched at 30°C were identical to and in the same order as those obtained from the analysis of the genes up-regulated =four-fold at 30°C (Table 1). At 37°C only seven out of the 10 pathways associated with the differentially expressed genes are identical to those from the GO enriched sequences. The three additional enriched pathways were histidine metabolism, butanoate metabolism, and phenylalanine metabolism.
![]()
Table 1: Top ten KEGG pathways associated with differentially expressed or GO
enriched genes.
View Table 1
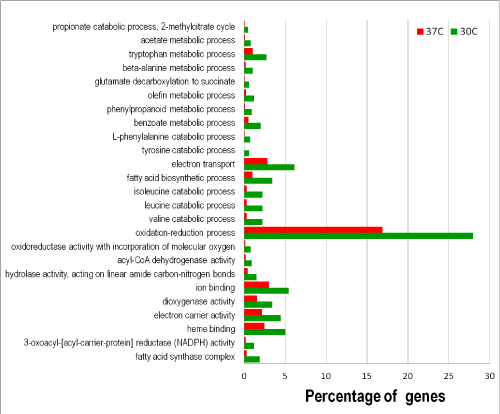 Figure 3: Functional categories of differentially expressed genes at 30°C and 37°C.
Figure 3: Functional categories of differentially expressed genes at 30°C and 37°C.
Gene up-regulated = four-fold was the selection criterion for differential expression. Fisher's Exact Test with the Benjamini and Hochberg FDR (False
Discovery Rate) correction at the significance threshold of 0.05 was used to obtain the adjusted p-values between the up-regulated test gene group and the
reference gene group.
View Figure 3
Discussion
Our results suggest that the constructed A. flavus GO database is useful for providing reasonable interpretations of the test RNA-Seq dataset. The GO enrichment analysis implies that A. flavus growth at 30°C favors degradation of amino acids mainly with nonpolar side chains, such as valine, leucine, isoleucine, phenylalanine, and tryptophan (Figure 3). In contrast, growth at 37°C favors the degradation of arginine and histidine (basic amino acids), aspartate and glutamate (acidic), and serine and threonine (uncharged but polar) (Table 1). The KEGG pathway analyses also confirm that 30°C promotes the degradation of the aforementioned nonpolar amino acids, while the higher growth temperature of 37°C enhances the biosynthesis of nonpolar amino acids. Therefore, the growth temperatures influence catabolism and anabolism of amino acids in A. flavus differently. A higher activity of carbohydrate and sucrose metabolism is found to be associated with up-regulated genes at 37°C, but the GO enrichment result, which is based on the highly selective criterion of FDR < 0.05, shows otherwise. Carbohydrate and sucrose metabolism ranks second in the whole-genome reference pathways (Table S1); thus the higher pathway activity observed in the differentially expressed gene population likely is insignificant. This may also be true for the pathway activities of purine metabolism and tryptophan metabolism, which rank first and seventh in the reference pathways (Tables 1 and (Supplemental Material), found at 37°C.
At each growth temperature, metabolically linked pathways are simultaneously influenced. For example, butanoate metabolism links to fatty acid degradation (see map at http://www.genome.jp/kegg/pathway/map/map00650.html). Propanoate metabolism links to degradation of valine, leucine, isoleucine and alanine (http://www.genome.jp/kegg-bin/show_pathway?map00640). These four pathways are actively operating at 30°C. Similarly, valine, leucine and isoleucine biosynthesis links to glycine, serine and threonine metabolism. Arginine and proline metabolism as well as histidine metabolism link to alanine, aspartate and glutamate metabolism. All of these pathways are actively operating at 37°C.
The largest class of volatile compounds produced by A. parasiticus grown at 30°C is derived from catabolic intermediates of leucine, isoleucine, and valine [22]. The pathway of valine, leucine and isoleucine degradation is active in A. flavus at 30°C, which suggests that degradation of these amino acids occurs preferably at 30°C in aspergilli. On the other hand, biosynthesis of these amino acids is active when A. flavus grows at 37°C (Table 1).
Aspergillus nidulans in response to hypoxia synthesizes de novo branched-chain amino acids of leucine, isoleucine, and valine [23]. Whether growth of A. flavus at suboptimal 37°C causes deprivation of adequate oxygen supply and results in hypoxic conditions is unclear. Phenylalanine metabolism (http://www.genome.jp/kegg/pathway/map/map00360.html) produces pyruvate, acetyl-CoA, fumarate, succinate, and succinyl-CoA. Tryptophan metabolism (http://www.genome.jp/kegg-bin/show_pathway?map00380+C000240) leads to acetyl-CoA production. Tyrosine metabolism (http://www.genome.jp/kegg/pathway/map/map00350.html) produces pyruvate, fumarate and succinate. These intermediates can enter the Tricarboxylic Acid (TCA) cycle (the citric acid/Krebs cycle) and may in part be involved in energy (ATP) production when A. flavus grows at 30°C.
Compared to those at 37°C the majority of pathways with higher activities at 30°C are involved in the production of acetyl-CoA. Acetyl-CoA has two principle fates: it either enters the TCA cycle to generate more ATP or it is used to synthesize new fatty acids. Many aspergilli contain high amounts of unsaturated fatty acids [18,24,25]. In A. flavus more than 70% of fatty acids in the total fatty acid content are unsaturated fatty acids, which are composed mainly of oleic acid (C18:1) and linoleic acid (C18:2) [18]. The observation that pathways of (saturated) fatty acid biosynthesis and degradation as well as biosynthesis of unsaturated fatty acids are active at 30°C may indicate the necessity of maintaining the high content of unsaturated fatty acids in A. flavus at the optimal 30°C growth temperature. The finding that the main cellular component involved at 30°C growth is the fatty acid synthase complex also supports this notion. An increase in fatty acid unsaturation has been well known for many fungi at lower growth temperatures [25-27].
Of the annotated A. flavus NRRL3357 genes in the current genome database at NCBI, JCVI, and Broad Institute 5,613 encode either conserved hypothetical proteins or hypothetical proteins. Among these genes 1,115 (20%) were further mapped and annotated with GO terms in this study, which gives a final total of 8,987 annotated genes. Therefore, 67% (8,987/13,485) of the total A. flavus genes can be used in functional genomics studies. Although this number is far from complete, it is an intrinsic drawback of all current GO databases. However, the percentage of annotated genes in the A. flavus GO database is in the range of genes annotated with GO terms including those of rice, sweet potato, hazelnut and salamander [28-31]. Besides the genes encoding conserved and/or hypothetical proteins, an additional 1,025 novel A. flavus genes have been identified from assembled RNA-Seq transcript data [16]. For A. oryzae, which is closely related to A. flavus, 1,116 novel transcripts are also reported [32]. The actual gene number of A. flavus likely is greater than originally predicted. Future inclusion of these genes if annotated will substantially increase the fidelity of the results for A. flavus functional genomics studies.
Online construction of a GO database is time-consuming. The blast, mapping and annotation steps in this work took well over 550 CPU hours. When necessary the current database can be updated periodically as more information about those unannotated genes is obtained. This can be achieved by individual or collaborating research groups to divide the genes to be annotated into manageable portions for generating GO sub-databases. These sub-databases then can be merged into the current database by using the "import annotation file" function of Blast2GO as an updated annotation file. This would greatly reduce the processing time in comparison to constructing an entirely new GO database. Despite nucleotide sequence variations in orthologs of related Aspergillus spices, such as A. parasiticus, A. nomius, A. bombycis, A. minisclerotigenes, A. arachidicola, A. pseudonomius, A. sojae, and A. oryzae [33,34], the encoded proteins should yield the same GO terms. GO databases for these aspergilli can now be constructed based on the current A. flavus GO database. The A. flavus orthologs can first be filtered out by using the NCBI BLAST+ tool [35]; only genes unique to each species need to be further annotated as indicated above. In conclusion, the A. flavus GO database constructed in this study should help to expedite the progress of functional genomics of A. flavus and related aspergilli.
References
-
Krishnan S, Manavathu EK, Chandrasekar PH (2009) Aspergillus flavus: an emerging non-fumigatus Aspergillus species of significance. Mycoses 52: 206-222.
-
Payne GA, Nierman WC, Wortman JR, Pritchard BL, Brown D et al. (2006) Whole genome comparison of Aspergillus flavus and A. oryzae. Med Mycol 44: 9-11.
-
Gibbons JG, Salichos L, Slot JC, Rinker DC, McGary KL, et al. (2012) The evolutionary imprint of domestication on genome variation and function of the filamentous fungus Aspergillus oryzae. Curr Biol 22: 1403-1409.
-
Machida M, Terabayashi Y, Sano M, Yamane N, Tamano K, et al. (2008) Genomics of industrial Aspergilli and comparison with toxigenic relatives. Food Addit Contam Part A Chem Anal Control Expo Risk Assess 25: 1147-1151.
-
Chang P-K, Wilkinson JR, Horn BW, Yu J, Bhatnagar D et al. (2007) Genes differentially expressed by Aspergillus flavus strains after loss of aflatoxin production by serial transfers. Appl Microbiol Biotechnol 77: 917-925.
-
Cary JW, OBrian GR, Nielsen DM, Nierman W, Harris-Coward P, et al. (2007) Elucidation of veA-dependent genes associated with aflatoxin and sclerotial production in Aspergillus flavus by functional genomics. Appl Microbiol Biotechnol 76: 1107-1118.
-
Wilkinson JR, Yu J, Bland JM, Nierman WC, Bhatnagar D et al. (2007) Amino acid supplementation reveals differential regulation of aflatoxin biosynthesis in Aspergillus flavus NRRL 3357 and Aspergillus parasiticus SRRC 143. Appl Microbiol Biotechnol 74: 1308-1319.
-
Reverberi M, Punelli M, Scala V, Scarpari M, Uva P, et al. (2013) Genotypic and phenotypic versatility of Aspergillus flavus during maize exploitation. PLoS One 8: e68735.
-
Dolezal AL, Obrian GR, Nielsen DM, Woloshuk CP, Boston RS, et al. (2013) Localization, morphology and transcriptional profile of Aspergillus flavus during seed colonization. Mol Plant Pathol 14: 898-909.
-
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621-628.
-
Conesa A, Götz S (2008) Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008: 619832.
-
Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44-57.
-
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, et al. (2009) ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25: 1091-1093.
-
Du Z, Zhou X, Ling Y, Zhang Z, Su Z (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38: W64-70.
-
Priebe S, Linde J, Albrecht D, Guthke R, Brakhage AA (2011) FungiFun: a web-based application for functional categorization of fungal genes and proteins. Fungal Genet Biol 48: 353-358.
-
Lin JQ, Zhao XX, Zhi QQ, Zhao M, He ZM (2013) Transcriptomic profiling of Aspergillus flavus in response to 5-azacytidine. Fungal Genet Biol 56: 78-86.
-
Yu J, Fedorova ND, Montalbano BG, Bhatnagar D, Cleveland TE, et al. (2011) Tight control of mycotoxin biosynthesis gene expression in Aspergillus flavus by temperature as revealed by RNA-Seq. FEMS Microbiol Lett 322: 145-149.
-
Fraga ME, Santana DM, Gatti MJ, Direito GM, Cavaglieri LR, et al. (2008) Characterization of Aspergillus species based on fatty acid profiles. Mem Inst Oswaldo Cruz 103: 540-544.
-
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, et al. (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36: 3420-3435.
-
Blüthgen N, Brand K, Cajavec B, Swat M, Herzel H, et al. (2005) Biological profiling of gene groups utilizing Gene Ontology. Genome Inform 16: 106-115.
-
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40: D109-114.
-
Roze LV, Chanda A, Laivenieks M, Beaudry RM, Artymovich KA et al. (2010) Volatile profiling reveals intracellular metabolic changes in Aspergillus parasiticus: veA regulates branched chain amino acid and ethanol metabolism. BMC Biochem 11: 33.
-
Shimizu M, Fujii T, Masuo S, Takaya N (2010) Mechanism of de novo branched-chain amino acid synthesis as an alternative electron sink in hypoxic Aspergillus nidulans cells. Appl Environ Microbiol 76: 1507-1515.
-
Stahl PD, Klug MJ (1996) Characterization and differentiation of filamentous fungi based on Fatty Acid composition. Appl Environ Microbiol 62: 4136-4146.
-
Suutari M (1995) Effect of growth temperature on lipid fatty acids of four fungi (Aspergillus niger, Neurospora crassa, Penicillium chrysogenum, and Trichoderma reesei). Arch Microbiol 164: 212-216.
-
Cheawchanlertfa P, Cheevadhanarak S, Tanticharoen M, Maresca B, Laoteng K (2011) Up-regulated expression of desaturase genes of Mucor rouxii in response to low temperature associates with pre-existing cellular fatty acid constituents. Mol Biol Rep 38: 3455-3462.
-
Duan C-H, Riley MB, Jeffers SN (2011) Effects of growth medium, incubation temperature, and mycelium age onproduction of five major fatty acids by six species of Phytophthora. Arch Phytopathol Plant Protect 44: 142-157.
-
Xu H, Gao Y, Wang J (2012) Transcriptomic analysis of rice (Oryza sativa) developing embryos using the RNA-Seq technique. PLoS One 7: e30646.
-
Che R, Sun Y, Wang R, Xu T (2014) Transcriptomic analysis of endangered Chinese salamander: identification of immune, sex and reproduction-related genes and genetic markers. PLoS One 9: e87940.
-
Ma H, Lu Z, Liu B, Qiu Q, Liu J (2013) Transcriptome analyses of a Chinese hazelnut species Corylus mandshurica. BMC Plant Biol 13: 152.
-
Tao X, Gu YH, Wang HY, Zheng W, Li X, et al. (2012) Digital gene expression analysis based on integrated de novo transcriptome assembly of sweet potato [Ipomoea batatas (L.) Lam]. PLoS One 7: e36234.
-
Wang B, Guo G, Wang C, Lin Y, Wang X, et al. (2010) Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res 38: 5075-5087.
-
Varga J, Frisvad JC, Samson RA (2011) Two new aflatoxin producing species, and an overview of Aspergillus section Flavi. Stud Mycol 69: 57-80.
-
Pildain MB, Frisvad JC, Vaamonde G, Cabral D, Varga J, et al. (2008) Two novel aflatoxin-producing Aspergillus species from Argentinean peanuts. Int J Syst Evol Microbiol 58: 725-735.
-
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, et al. (2009) BLAST+: architecture and applications. BMC Bioinformatics 10: 421.
