
Journal of Musculoskeletal Disorders and Treatment
Developmental Insights into Osteoarthritis Increase the Applicability of New Animal Models
Brunt LH1, Kague E2 and Hammond CL1*
1Department of Physiology, Pharmacology and Neuroscience, Biomedical Sciences, University of Bristol, UK
2Department of Genetics and Evolutionary Biology, University of São Paulo, Brazil
*Corresponding author:
Hammond CL, Department of Physiology, Pharmacology and Neuroscience, Biomedical Sciences, University of Bristol, UK, Tel: 0044-0-117-331-2228, E-mail: Chrissy.hammond@bristol.ac.uk
J Musculoskelet Disord Treat, JMDT-2-017, (Volume 2, Issue 3), Review Article
Received: March 30, 2016: Accepted: July 11, 2016: Published: July 14, 2016
Citation: Brunt LH, Kague E, Hammond CL (2016) Developmental Insights into Osteoarthritis Increase the Applicability of New Animal Models. J Musculoskelet Disord Treat 2:017.
Copyright: © 2016 Brunt LH, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Osteoarthritis (OA) is an increasingly common degenerative joint condition, estimated to affect more than 100 million people worldwide and more than 40% of people over 70 years of age. There is currently no pharmacological cure. The genetic contribution to osteoarthritis is estimated at between 39 and 60% in knee and hip OA respectively and a number of Genome Wide association studies have identified a number of alleles and loci that confer increased susceptibility to OA. In the last few years a number of key studies have demonstrated that genes known to play a role in the normal skeletal developmental processes of endochondral ossification, chondrocyte hypertrophy and joint formation are reactivated during osteoarthritis progression. Significantly, in some cases modification of these developmental genes can modulate osteoarthritis severity.
The high levels of genetic contribution make osteoarthritis an ideal target for both forward genetic screening efforts focused on identifying novel genes involved in chondrocyte differentiation and hypertrophy, which are likely to be involved in osteoarthritis progression and reverse genetics focused on modulation of genes involved in developmental skeletogenesis in later life. The functional study of genes associated with aging can be problematic in part because of the limited availability of animal models and the high costs associated with keeping aged animals. Therefore, the developmental aspect opens up the prospect of using chemical screens, such as those commonly performed in the zebrafish and frogs, to identify potential pharmaceutical modifiers of disease phenotypes. Here, we explore the current models for OA and compare genes, including mechanosensitive genes, involved in developmental joint formation and osteoarthritis.
Keywords
Osteoarthritis, Development, Chondrocyte hypertrophy, Endochondral ossification, Genetics, Joint morphogenesis, Animal models
A Brief Introduction to Osteoarthritis
Osteoarthritis is the most common form of arthritis, and represents a major cause of disabilities in the western world [1]. Current treatment for OA includes pain management and in more severe cases surgical replacement of the joint, known as total joint replacement (TJR) is performed [2]. However, a pharmaceutical cure to reverse the damage caused by OA has not yet been found.
Osteoarthritis is a disease of the whole joint, involving changes to many of the joint tissues. These include significant loss of the articular cartilage, which normally covers the bone surfaces, through breakdown of the structural proteins of the cartilage matrix [3]. This loss of cartilage is twinned with changes to the underlying bone, primarily thickening of the subchondral bone and the formation of osteophytes, which can protrude into the joint causing stiffness [4] and an inflammatory response mediated principally by proinflammatory cytokines [5].
Much is known about the environmental factors that increase OA susceptibility, principally age, sports injury, obesity and diet [6,7]. However, in recent years there has been a shift in attention from studies focused primarily on these environmental effects on cartilage destruction, towards the genetic factors regulating disease onset. It has been estimated from a number of twin studies that the genetic contribution to OA lies between 39 and 60% in hip and knee OA, respectively [8,9]. One aspect that makes large scale genetic epidemiology studies of OA somewhat difficult is the use of an unambiguous disease phenotype, as the disease is highly variable ranging from symptom-free radiographic disease to painful end stage OA requiring joint replacement surgery (reviewed in [10]). The largest and most extensive Genome Wide Association Study (GWAS) published to date was the arcOGEN study, which included 7400 OA cases and identified a number of loci [11]. Since then many other association studies have been published (with varying power) which have identified further risk alleles in a number of other genes [12-14]. Genes involved in early stages of disease pathogenesis are likely to present the best targets for development of new therapies; therefore their identification is of paramount importance.
It has long been appreciated that one of the most damaging aspects of OA is the body's own vigorous attempt to repair the damage to articular cartilage [15]. Cartilage repair recapitulates a number of developmental pathways, including those involved in the normal embryonic replacement of cartilage with bone during endochondral ossification [16,17]. Of note, there is an increasing amount of evidence that aberrant reactivation of embryonic skeletal processes might play a contributory factor in disease progression. In this review we show recent evidence that reactivation of pathways relevant to skeletal development and joint morphogenesis contribute to OA risk, and discuss how this might open up new avenues for therapeutic intervention. Given that animal models of OA are of great value in addressing these issues, we also summarize the advantages and limitations of the different models.
Endochondral ossification and osteoarthritis
Recently, a number of groups have identified a set of genes with a central role in OA progression and pathogenesis (summarised in Table 1), and these genes share well-characterised roles in developmental endochondral ossification; spanning a number of types, from signalling proteins like Gdf5, hormones such as PTHrP, transcription factors such as Sox9, ERG and structural matrix proteins such as Type X collagen, amongst others. Together these reports suggest the involvement of a number of important and well known signalling pathways such as the Hedgehog, Wnt, BMP and TGFbeta pathways in influencing OA susceptibility and disease progression. Many of the genes (summarised in Table 1 and in Figure 1) appear to exert their effects in the disease by promoting the transition from a quiescent state to the re-entry of the chondrocyte into the cell cycle and differentiation to a hypertrophic state, similar to their role in the control of developmental endochondral ossification. A number of these genes appear to play a role in the 'early' stages of OA, raising the tantalising prospect that amongst the proteins controlling the entry to chondrocyte hypertrophy may be pharmaceutical drug targets that could be manipulated to limit OA progression. Supporting this idea, modulation of some of these 'developmental' genes has been demonstrated to decrease the severity of OA in vivo [18]. Indeed many new OA drugs currently in preclinical studies or in early stages of human trials modulate the developmental pathways discussed above; reviewed in [2].
.
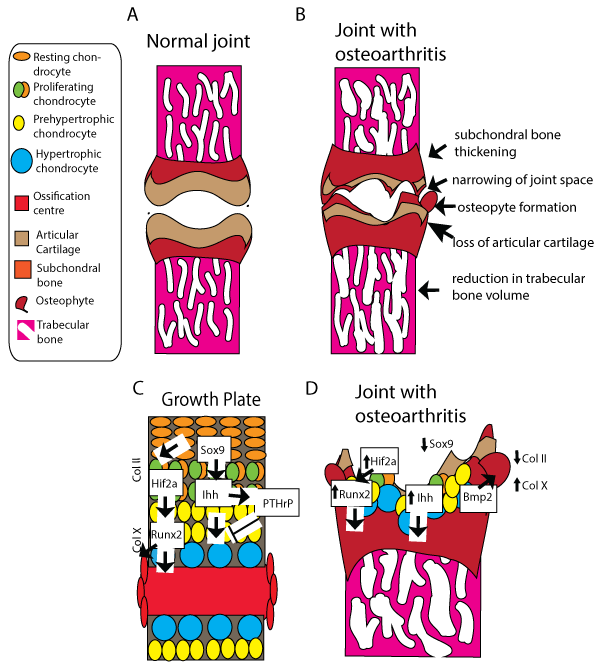
Figure 1: Osteoarthritis phenotype vs. endochondral ossification –changes and markers common to both processes.
A: Normal joints feature intact articular cartilage covering subchondral and trabecular bone; B: Osteoarthritic joint features include thinned articular cartilage, exposed subchondral bone, osteophytes formation and trabecular thinning; C: Schematic of a cartilage growth plate undergoing endochondral ossification from the resting chondrocytes to the ossification centre. Cell types/stages are referenced in the key. Molecular regulators and markers of stages of endochondral ossification. Activation arrowheads and inhibitory arrows represent diffusion of signals to site of action; D: Molecular changes related to osteoarthritis. Large arrows represent diffusion of signals to site of action. Small arrows represent up and down regulation of expression in osteoarthritis.
View Figure 1
![]()
Table 1: Comparative genetics of endochondral ossification/osteoarthritis.
View Table 1
This re-employment of developmental transcription factors and return to the 'fetal gene program' is a mechanism common to number of pathological states of the heart [19,20] and the brain [21].
Traditional models of OA: In vitro models, stem cells, tissue engineering and in vivo models
Currently the majority of research focused on OA is performed using either in vitro models (such as primary cultures of normal and osteoarthritic chondrocytes), or using various rodent models, predominantly in mouse, rat or guinea pig.
Since chondrocytes constitute the main cell type in a normal adult joint, cell cultures of chondrocytes are commonly used. These can either be monolayer cultures [22,23], 3D cultures [24], or cultures on artificial substrates [25]. They can be derived either from chondrocyte cells lines or cultured from stem cells such as mesenchymal stem cells (MSCs), which are often taken from patients carrying specific risk alleles to test the effects on chondrogenesis [26]. More recently, induced pluripotent stem cells have been used [27]. Additionally, explants from laboratory animals, such as mouse ribcage explants or calvaria have been used to study the effects of known OA-inducing stimuli (e.g. levels of mechanical stress, inflammation) in vitro [28]. Culture systems and cartilage tissue engineering are reviewed in detail elsewhere [29] and will not be covered further here.
Many animal models traditionally used to study OA have focused on destruction of the cartilage in weight bearing joints. This is usually achieved either through surgery by disrupting the medial meniscus, by destabilising the whole joint by anterior cruciate ligament transection, or by destruction of cartilage through chemical means, such as injection of collagenases or papain [30]. The extent of OA progression is then characterised by histomorphological studies, applying a set of defined criteria, which has been set out for each animal species by OARSI (Osteoarthritis Research Society International) to allow closer correlation between the results obtained from animal studies to human clinical characteristics [31-34].
One problem with surgical models, while undoubtedly of value, is that by focusing mainly on cartilage destruction they may not give much insight into subtle changes occurring during the early stages of OA and often do not fully recapitulate the transcriptional changes which are observed between human normal and osteoarthritic cartilage [4]. They, therefore, risk missing some of the changes occurring in the earliest and potentially most treatable stages of OA.
In addition to surgical models, an increasing number of genetically modified mouse models are established, in which genes identified from in vitro, human genetic, or other animal research models are knocked out in a targeted fashion, then analysed for joint phenotypes and OA. These include such mouse knockouts as Type II and Type IX collagen [35,36], COMP (Tsp5) [37], CCN3/Nov [38] and Ephrin B2 [39]. While these rodent mutants allow OA onset to be studied, it remains difficult to follow disease progression at a cellular level, as dynamic imaging of the skeleton at cellular resolution is impossible. As such imaging tends to focus on radiography and MRI in live animals twinned with post mortem histology. Therefore, ideally new models will combine the ability to identify novel genes with functional genomics and biochemical data and on the mechanisms by which these genes exert their effects and ideally the ability to use dynamic imaging. The developmental angle to the disease raises the tantalising prospect of using additional models to complement those currently used for OA research such as zebrafish, medaka and xenopus.
The uncovering of a 'developmental' aspect of OA should facilitate use of traditional developmental models such as mice and zebrafish, which are ideally suited for functional genetic studies. These include phenotype-driven forward genetic screens to identify novel genes involved in OA, and offer the advantage of an unbiased approach to identifying new genes through randomly generated mutagenesis approaches. ENU-based (N-ethyl-N-nitroso urea) random mutagenesis forward genetic screens in mouse are increasingly being used [40]. Through one such screen a new allele of Gdf5 was identified, the mutant displaying joint malformation and early onset of OA [69]. Finding back a gene already implicated in OA pathogenesis provides a proof of principal that forward genetic screens can identify OA susceptibility genes. Forward genetic screens have been used successfully for 20 years in zebrafish [70,71]. They have only more recently been applied to skeletal research, but are already beginning to identify genes involved in skeletogenesis [72-76]. The zebrafish mutant's dackel (dak) and pinscher (pic), have identified genes involved in the human bone disease Hereditary Multiple Exostoses (HME) [77], and further insight into osteogenesis imperfecta has come from zebrafish [78]. Furthermore, ageing zebrafish has been shown to develop skeletal phenotypes that strongly resemble human osteoarthritis [79]. Therefore, while zebrafish form fewer synovial joints (they only have synovial joints in the craniofacial skeleton), they have the potential to complement existing small animal models in understanding the genetic link between skeletogenesis and osteoarthritis.
While forward genetic screening provides the potential of identification of new genes related to disease, reverse genetics is a useful tool for functional studies of known genes, for example those identified from GWAs studies that require further functional analyses. Site-specific genome editing has become easier and suitable over the years. Traditional technology of Cre/loxp highly used in mice has enriched the knowledge on bone formation and also OA with the generation of conditional knockout animal models [80] but also transgenic lines for lineage tracing in mice and live zebrafish [81-85]. Lineage tracing experiments have contributed to the identification of different populations of osteoprogenitor cells in hypertrophic cartilage and origins of heterotopic ossifications, helping to understand the “developmental” aspect of OA and reasons why cartilage elements might respond differentially to treatments [86]. New technologies of genome editing have emerged, ZFN (Zinc Finger Nucleases) and TALEN (Transcription Activator-Like Effector Nuclease) [87] have been used elsewhere for functional tests in a variety of model organisms, for example frame shift alleles for kif6 were generated in zebrafish through TALEN and confirmed its role in development of spinal scoliosis [88]. More recently the advent of Crispr (Clustered Regularly- Interspaced Short Palindromic Repeats) has revolutionized the field providing a powerful facility over other methods to edit the genome. The system has been used successfully in a diversity of organisms including mice, rat, zebrafish, other vertebrates and cell lines (stable, iPSCs and primary) to either generate knockouts, tissue specific gene disruption, deletion of regulatory regions or correction of single base pair mutations [89-93]. The technology has yet to be extensively used for the study of bone formation and related diseases, however Crispr/Cas9 has already contributed to attribute function to upstream regions of Mmp13 (Matrix metalloproteinase 13) that when deleted result in complete loss of basal transcript activity [94]. Crispr has also been used to generate stable rat chondrocyte Aggrecan knockout cell lines [95], and to study Lrp5 during cell migration and shaping of the craniofacial skeleton [96]. The possibility of correction of the causative mutations may in future be applied to treatment of a spectrum of diseases including OA. Furthermore, multi target Crispr/cas9 combined with faster platforms such as zebrafish has the potential to validate candidate genes previously identified through GWAS and elucidate the importance of groups of altered genes for the cause of complex diseases as OA.
The generation of an increasing number of transgenic lines marking chondrocytes, osteoblasts and osteoclasts via promoters such as osterix [97-99], osteocalcin and runx2 [100,101], or col2a1 and col10a1 [102,103] along with those reporting on signalling pathway activity [104] will further benefit fish skeletal research. The increasing use of zebrafish and Xenopus for skeletal research will likely include screening of chemical libraries [105,106]. This could be through classical screening techniques on the many fish skeletal transgenic lines. Additionally, ex vivo approaches such as screening for skeletal compounds on scales removed from transgenic zebrafish expressing an sp7 (osterix) luciferase transgene have already identified a number of novel osteogenic compounds [107].
Joint shape, foetal movement and biomechanics
Loading, wear and tear and biomechanics have long been understood to play a role in OA pathogenesis, and it may be that there is a developmental aspect to the role of joint biomechanics in OA pathogenesis (see Figure 2 for genes in common between both joint development and OA). We now appreciate that the mechanical environment experienced during early development is important for normal skeletal development and correct joint morphogenesis. There are a number of conditions and syndromes for which abnormal or reduced movement are believed to be causal; including the relatively common developmental dysplasia of the hip (DDH sometimes also known as congenital dysplasia of the hip or CDH), which affects between 1.3 per 1000 births [108], arthrogryposis affecting 1:4000 births [109] and fetal akinesia deformation sequence (FADS) in which fetal movements are completely lacking, leading to joint contractures, craniofacial deformities and growth restriction, which affects 1:15000 births [110,111].
.
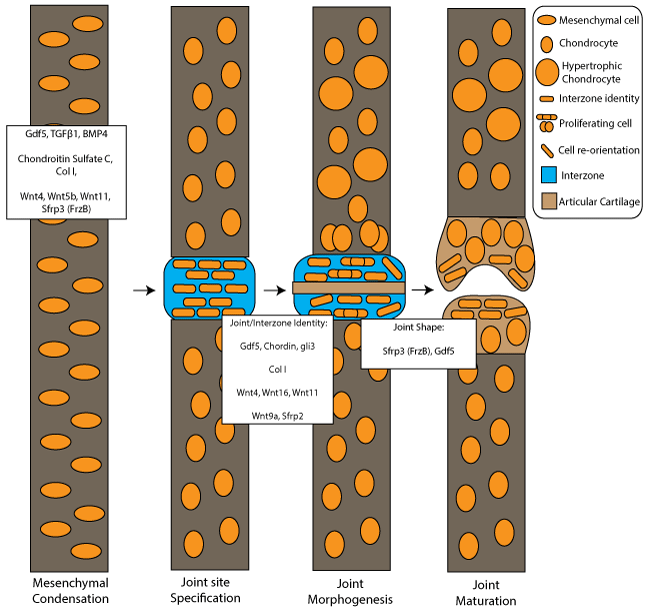
Figure 2: Osteoarthritis-related genes involved in joint development from mesenchymal condensation to joint maturation.
Joint development stages include mesenchymal condensation, joint site specification at the interzone region, joint morphogenesis and joint maturation. White boxes show osteoarthritis–related genes associated with mesenchymal condensation, joint and interzone identity and those implicated with joint shape. The key denotes cell/tissue types and cell behaviour changes during joint morphogenesis.
View Figure 2
Genes and joint shape
From developmental studies in animal models, it is evident that mechanical forces from muscle activity are required for accurate joint formation [112,113]. The identification of mechanosensitive genes and the roles they play at the developing joint are starting to be elucidated from regulation of interzone cell differentiation, aiding cavitation and promoting chondrogenesis and morphogenesis [114-116]. The Wnt pathway has been implicated as mechanosensitive with 34 Wnt genes differentially expressed between control and muscle-less Splotch mutant mice in the developing humerus, [117]. Beta-catenin, which acts downstream of the canonical Wnt pathway, also showed reduced activation at the joint site in mice Splotch mutants and local loss of Wnt9a expression, [118]. Interestingly, the Wnt pathway has been implicated as a major player in OA progression [119]. As a result of muscle paralysis during early development many genes relevant to OA, such as Col2a1, BMP2, and PTHrP, show altered expression at the developing knee joint, [116]. Jaw movement constraint in ex utero mice resulted in reduction of Ihh and PTHrP expression [114]. Gdf5, which is normally down regulated prior to cavitation, was reduced in cultured articular surface cells following the application of mechanical strain [115]. Immobilisation lead to elevation of Gdf5 in fused joints, reflecting the increase in numbers of chondrocytes at the joint site that express Gdf5 for chondrogenesis. Splotch mice, however, displayed an initial maintenance, and then decreased expression of Gdf5 at the interzone as a result of the absence of mechanical strain, [118]. The identification of genes that are misexpressed at the joint in animal models following reduced muscle activity can lead to further opportunities to identify the role that these mechanosensitive genes play in OA.
Locating regions that are under strain during development can help pin point those cells which are most likely to be responding to biomechanical cues to identify novel mechanosensitive genes and pathways. Finite Element (FE) modelling is an engineering technique that can computationally calculate and map the magnitude and location of strains acting on a 3d model [120]. FE analysis (FEA) has been exploited to model strains acting on skeletal elements during bone formation [121], the pattern of strains present during chick knee joint and zebrafish jaw joint morphogenesis [122,123]. Areas of high strain have been found to occur at regions that normally undergo cell behaviour changes such as proliferation or cell reorientation that impact on joint shape and that are affected in immobilised models [122,123]. FE modelling can be used to predict changes to strain patterns as a result of reduced muscle activity and can be used to identify mechanosensitive genes, such as ColX and Ihh involved in bone formation [124]. Further studies could help identify more mechanosensitive genes involved in joint morphogenesis in development that play a role during OA.
Future of OA research and potential for use of new models/screens
As outlined above, recent genetic findings have identified a number of osteoarthritis susceptibility genes which also regulate Endochondral ossification and joint morphogenesis during skeletal morphogenesis; variation in these genes seems likely to be involved both in the onset of osteoarthritis and subsequent progression towards clinical outcomes. This developmental focus will allow a number of powerful models currently used to study developmental genetics and cell behaviour including chick, xenopus and zebrafish to be brought into osteoarthritis research, to complement existing research tools. These include forward genetic screens, reverse genetics with CRISPr and high-throughput in vivo chemical screens, to identify and functionally characterise new targets for pharmaceutical intervention and novel chemical modulators of pathways. This heralds an exciting period for OA research and hopefully new therapies for this disease will soon follow.
Acknowledgments
CLH and EK would like to thank Arthritis Research UK for funding through grants 19476 and 21211.
References
-
Dieppe PA, Lohmander LS (2005) Pathogenesis and management of pain in osteoarthritis. Lancet 365: 965-973.
-
Zhang W, Ouyang H, Dass CR, Xu J (2016) Current research on pharmacologic and regenerative therapies for osteoarthritis. Bone Res 4: 15040.
-
Heinegård D, Saxne T (2011) The role of the cartilage matrix in osteoarthritis. Nat Rev Rheumatol 7: 50-56.
-
Little CB, Fosang AJ (2010) Is cartilage matrix breakdown an appropriate therapeutic target in osteoarthritis--insights from studies of aggrecan and collagen proteolysis? Curr Drug Targets 11: 561-575.
-
Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H (2011) Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol 7: 33-42.
-
Li S, Micheletti R (2011) Role of diet in rheumatic disease. Rheum Dis Clin North Am 37: 119-133.
-
Zhang Y, Jordan JM (2010) Epidemiology of osteoarthritis. Clin Geriatr Med 26: 355-369.
-
Lanyon P, Muir K, Doherty S, Doherty M (2000) Assessment of a genetic contribution to osteoarthritis of the hip: sibling study. BMJ 321: 1179-1183.
-
Zhai G, Hart DJ, Kato BS, MacGregor A, Spector TD (2007) Genetic influence on the progression of radiographic knee osteoarthritis: a longitudinal twin study. Osteoarthritis Cartilage 15: 222-225.
-
Loughlin J (2015) Genetic contribution to osteoarthritis development: current state of evidence. Curr Opin Rheumatol 27: 284-288.
-
arcOGEN Consortium, arcOGEN Collaborators, Zeggini E, Panoutsopoulou K, Southam L, Rayner NW, et al. (2012) Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome-wide association study. Lancet 380: 815-823.
-
Evangelou E, Kerkhof HJ, Styrkarsdottir U, Ntzani EE, Bos SD, et al. (2014) A meta-analysis of genome-wide association studies identifies novel variants associated with osteoarthritis of the hip. Ann Rheum Dis 73: 2130-2136.
-
Moon S, Keam B, Hwang MY, Lee Y, Park S, et al. (2015) A genome-wide association study of copy-number variation identifies putative loci associated with osteoarthritis in Koreans. BMC Musculoskelet Disord 16: 76.
-
Rodriguez-Fontenla C, Calaza M, Evangelou E, Valdes AM, Arden N, et al. (2014) Assessment of osteoarthritis candidate genes in a meta-analysis of nine genome-wide association studies. Arthritis Rheumatol 66: 940-949.
-
Harrison MH, Schajowicz F, Trueta J (1953) Osteoarthritis of the hip: a study of the nature and evolution of the disease. J Bone Joint Surg Br 35-35B: 598-626.
-
Onyekwelu I, Goldring MB, Hidaka C (2009) Chondrogenesis, joint formation, and articular cartilage regeneration. J Cell Biochem 107: 383-392.
-
Saito T, Fukai A, Mabuchi A, Ikeda T, Yano F, et al. (2010) Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat Med 16: 678-686.
-
Lin AC, Seeto BL, Bartoszko JM, Khoury MA, Whetstone H, et al. (2009) Modulating hedgehog signaling can attenuate the severity of osteoarthritis. Nat Med 15: 1421-1425.
-
Oka T, Xu J, Molkentin JD (2007) Re-employment of developmental transcription factors in adult heart disease. Semin Cell Dev Biol 18: 117-131.
-
Taegtmeyer H, Sen S, Vela D (2010) R0065turn to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci 1188: 191-198.
-
Kovacs GG, Adle-Biassette H, Milenkovic I, Cipriani S, van Scheppingen J5, et al. (2014) Linking pathways in the developing and aging brain with neurodegeneration. Neuroscience 269: 152-172.
-
Beekhuizen M, Bastiaansen-Jenniskens YM, Koevoet W, Saris DB, Dhert WJ, et al. (2011) Osteoarthritic synovial tissue inhibits proteoglycan production in human osteoarthritic cartilage; Establishment and characterisation of a long-term coculture. Arthritis Rheum 63: 1918-1927.
-
Chowdhury TT, Salter DM, Bader DL, Lee DA (2004) Integrin-mediated mechanotransduction processes in TGFbeta-stimulated monolayer-expanded chondrocytes. Biochem Biophys Res Commun 318: 873-881.
-
Chowdhury TT, Arghandawi S, Brand J, Akanji OO, Bader DL, et al. (2008) Dynamic compression counteracts IL-1beta induced inducible nitric oxide synthase and cyclo-oxygenase-2 expression in chondrocyte/agarose constructs. Arthritis Res Ther 10: R35.
-
Rankin KS, Lakey RL, Gerrand CH, Sprowson AP, McCaskie AW, et al. (2010) A novel in vitro model to investigate behavior of articular chondrocytes in osteoarthritis. J Rheumatol 37: 426-431.
-
Johnson K, Reynard LN, Loughlin J (2015) Functional characterisation of the osteoarthritis susceptibility locus at chromosome 6q14.1 marked by the polymorphism rs9350591. BMC Med Genet 16: 81.
-
Lietman SA (2016) Induced pluripotent stem cells in cartilage repair. World J Orthop 7: 149-155.
-
Gosset M, Berenbaum F, Levy A, Pigenet A, Thirion S, et al. (2006) Prostaglandin E2 synthesis in cartilage explants under compression: mPGES-1 is a mechanosensitive gene. Arthritis Res Ther 8: R135.
-
Mardones R, Jofré CM, Minguell JJ (2015) Cell Therapy and Tissue Engineering Approaches for Cartilage Repair and/or Regeneration. Int J Stem Cells 8: 48-53.
-
Ameye LG, Young MF (2006) Animal models of osteoarthritis: lessons learned while seeking the "Holy Grail". Curr Opin Rheumatol 18: 537-547.
-
Cook JL, Kuroki K, Visco D, Pelletier JP, Schulz L, et al. (2010) The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the dog. Osteoarthritis Cartilage 18: S66-79.
-
Gerwin N, Bendele AM, Glasson S, Carlson CS (2010) The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the rat. Osteoarthritis Cartilage 18: S24-34.
-
Glasson SS, Chambers MG, Van Den Berg WB, Little CB (2010) The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage S17-23.
-
Kraus VB, Huebner JL, DeGroot J, Bendele A (2010) The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the guinea pig. Osteoarthritis Cartilage 18: S35-52.
-
Kimura T, Nakata K, Tsumaki N, Miyamoto S, Matsui Y, et al. (1996) Progressive degeneration of articular cartilage and intervertebral discs. An experimental study in transgenic mice bearing a type IX collagen mutation. Int Orthop 20: 177-181.
-
Rintala M, Metsäranta M, Säämänen AM, Vuorio E, Rönning O (1997) Abnormal craniofacial growth and early mandibular osteoarthritis in mice harbouring a mutant type II collagen transgene. J Anat 190: 201-208.
-
Blumbach K, Niehoff A, Paulsson M, Zaucke F (2008) Ablation of collagen IX and COMP disrupts epiphyseal cartilage architecture. Matrix Biol 27: 306-318.
-
Roddy KA, Boulter CA (2015) Targeted mutation of NOV/CCN3 in mice disrupts joint homeostasis and causes osteoarthritis-like disease. Osteoarthritis Cartilage 23: 607-615.
-
Valverde-Franco G, Lussier B, Hum D, Wu J, Hamadjida A, et al. (2016) Cartilage-specific deletion of ephrin-B2 in mice results in early developmental defects and an osteoarthritis-like phenotype during aging in vivo. Arthritis Res Ther 18: 65.
-
Shen G (2005) The role of type X collagen in facilitating and regulating endochondral ossification of articular cartilage. Orthod Craniofac Res 8: 11-17.
-
von der Mark K, Kirsch T, Nerlich A, Kuss A, et al. (1992) Type X collagen synthesis in human osteoarthritic cartilage. Indication of chondrocyte hypertrophy. Arthritis Rheum 35: 806-811.
-
Yoshida T, Horiuchi T, Sakamoto H, Inoue H, Takayanagi H, et al. (1998) Production of parathyroid hormone-related peptide by synovial fibroblasts in human osteoarthritis. FEBS Lett 433: 331-334.
-
Kronenberg HM (2006) PTHrP and skeletal development. Ann N Y Acad Sci 1068: 1-13.
-
Kamekura S, Kawasaki Y, Hoshi K, Shimoaka T, Chikuda H, et al. (2006) Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum 54: 2462-2470.
-
Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, D, et al. (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89: 755-764.
-
Ueta C, Iwamoto M, Kanatani N, Yoshida C, Liu Y, et al. (2001) Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J Cell Biol 153: 87-100.
-
Tamiya H, Ikeda T, Jeong JH, Saito T, Yano F, et al. (2008) Analysis of the Runx2 promoter in osseous and non-osseous cells and identification of HIF2A as a potent transcription activator. Gene 416: 53-60.
-
Inada M, Wang Y, Byrne MH, Rahman MU, Miyaura C, et al. (2004) Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc Natl Acad Sci U S A 101: 17192-17197.
-
Mitchell PG, Magna HA, Reeves LM, Lopresti-Morrow LL, Yocum SA, et al. (1996) Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J Clin Invest 97: 761-768.
-
Bell DM, Leung KK, Wheatley SC, Ng LJ, Zhou S, et al. (1997) SOX9 directly regulates the type-II collagen gene. Nat Genet 16: 174-178.
-
Cucchiarini M, Thurn T, Weimer A, Kohn D, Terwilliger EF, et al. (2007) Restoration of the extracellular matrix in human osteoarthritic articular cartilage by overexpression of the transcription factor SOX9. Arthritis Rheum 56: 158-167.
-
Storm EE, Kingsley DM (1999) GDF5 coordinates bone and joint formation during digit development. Dev Biol 209: 11-27.
-
Valdes AM, Evangelou E, Kerkhof HJ, Tamm A, Doherty SA, et al. (2011) The GDF5 rs143383 polymorphism is associated with osteoarthritis of the knee with genome-wide statistical significance. Ann Rheum Dis 70: 873-875.
-
Enomoto-Iwamoto M, Kitagaki J, Koyama E, Tamamura Y, Wu C, et al. (2002) The Wnt antagonist Frzb-1 regulates chondrocyte maturation and long bone development during limb skeletogenesis. Dev Biol 251: 142-156.
-
Evangelou E, Chapman K, Meulenbelt I, Karassa FB, Loughlin J, et al. (2009) Large-scale analysis of association between GDF5 and FRZB variants and osteoarthritis of the hip, knee, and hand. Arthritis Rheum 60: 1710-1721.
-
Ramos YF, den Hollander W, Bovee JV, Bomer N, van der Breggen R, et al. (2014) Genes involved in the osteoarthritis process identified through genome wide expression analysis in articular cartilage; the RAAK study. PloS one 9.
-
Wada N, Kawakami Y, Ladher R, Francis-West PH, Nohno T (1999) Involvement of Frzb-1 in mesenchymal condensation and cartilage differentiation in the chick limb bud. Int J Dev Biol 43: 495-500.
-
Ehlen HW, Buelens LA, Vortkamp A (2006) Hedgehog signaling in skeletal development. Birth Defects Res C Embryo Today 78: 267-279.
-
Blaney Davidson EN, Vitters EL, van Beuningen HM, van de Loo FA, van den Berg WB, et al. (2007) Resemblance of osteophytes in experimental osteoarthritis to transforming growth factor beta-induced osteophytes: limited role of bone morphogenetic protein in early osteoarthritic osteophyte formation. Arthritis Rheum 56: 4065-4073.
-
Keller B, Yang T, Chen Y, Munivez E, Bertin T, et al. (2011) Interaction of TGFβ and BMP signaling pathways during chondrogenesis. PLoS One 6: e16421.
-
Otsuki S, Taniguchi N, Grogan SP, D'Lima D, Kinoshita M, et al. (2008) Expression of novel extracellular sulfatases Sulf-1 and Sulf-2 in normal and osteoarthritic articular cartilage. Arthritis Res Ther 10: R61.
-
Raine EV, Dodd AW, Reynard LN, Loughlin J (2013) Allelic expression analysis of the osteoarthritis susceptibility gene COL11A1 in human joint tissues. BMC Musculoskelet Disord 14: 85.
-
Xu L, Flahiff CM, Waldman BA, Wu D, Olsen BR, Setton, et al. (2003) Osteoarthritis-like changes and decreased mechanical function of articular cartilage in the joints of mice with the chondrodysplasia gene (cho). Arthritis Rheum 48: 2509-2518.
-
Hayes AJ, Tudor D, Nowell MA, Caterson B, Hughes CE (2008) Chondroitin sulfate sulfation motifs as putative biomarkers for isolation of articular cartilage progenitor cells. J Histochem Cytochem 56: 125-138.
-
Echtermeyer F, Bertrand J, Dreier R, Meinecke I, Neugebauer K, et al. (2009) Syndecan-4 regulates ADAMTS-5 activation and cartilage breakdown in osteoarthritis. Nat Med 15: 1072-1076.
-
Howard M, Tuan RS, Wallis GA (2016) The function and interrelationship between GDF5 and ERG-010 during chondrogenesis in vitro. In vitro cellular & developmental biology. Animal 52: 182-192.
-
Iwamoto M, Tamamura Y, Koyama E, Komori T, Takeshita N, et al. (2007) Transcription factor ERG and joint and articular cartilage formation during mouse limb and spine skeletogenesis. Dev Biol 305: 40-51.
-
Ohta Y, Okabe T, Larmour C, Di Rocco A, Maijenburg MW, et al. (2015) Articular cartilage endurance and resistance to osteoarthritic changes require transcription factor Erg. Arthritis Rheumatol 67: 2679-2690.
-
Stottmann RW, Beier DR (2010) Using ENU mutagenesis for phenotype-driven analysis of the mouse. Methods Enzymol 477: 329-348.
-
Masuya H, Nishida K, Furuichi T, Toki H, Nishimura G, et al. (2007) A novel dominant-negative mutation in Gdf5 generated by ENU mutagenesis impairs joint formation and causes osteoarthritis in mice. Hum Mol Genet 16: 2366-2375.
-
Driever W, Solnica-Krezel L, Schier AF, Neuhauss SC, Malicki J, et al. (1996) A genetic screen for mutations affecting embryogenesis in zebrafish. Development 123: 37-46.
-
Haffter P, Granato M, Brand M, Mullins MC, Hammerschmidt M, et al. (1996) The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development 123: 1-36.
-
Huitema LF, Apschner A, Logister I, Spoorendonk KM, Bussmann J, et al. (2012) Entpd5 is essential for skeletal mineralization and regulates phosphate homeostasis in zebrafish. Proc Natl Acad Sci U S A 109: 21372-21377.
-
Spoorendonk KM, Hammond CL, Huitema LFA, Vanoevelen J, Schulte-Merker S (2010) Zebrafish as a unique model system in bone research: the power of genetics and in vivo imaging. J Appl Ichthyol 26: 219-224.
-
Spoorendonk KM, Peterson-Maduro J, Renn J, Trowe T, Kranenbarg S, et al. (2008) Retinoic acid and Cyp26b1 are critical regulators of osteogenesis in the axial skeleton. Development 135: 3765-3774.
-
Vanoevelen J, Janssens A, Huitema LF, Hammond CL, Metz JR, et al. (2011) Trpv5/6 is vital for epithelial calcium uptake and bone formation. FASEB J 25: 3197-3207.
-
Clément A, Wiweger M, von der Hardt S, Rusch MA, Selleck SB, et al. (2008) Regulation of zebrafish skeletogenesis by ext2/dackel and papst1/pinscher. PLoS Genet 4: e1000136.
-
Fisher S, Jagadeeswaran P, Halpern ME (2003) Radiographic analysis of zebrafish skeletal defects. Dev Biol 264: 64-76.
-
Hayes AJ, Reynolds S, Nowell MA, Meakin LB, Habicher J, et al. (2013) Spinal deformity in aged zebrafish is accompanied by degenerative changes to their vertebrae that resemble osteoarthritis. PLoS One 8: e75787.
-
Vasheghani F, Monemdjou R, Fahmi H, Zhang Y, Perez G, et al. (2013) Adult cartilage-specific peroxisome proliferator-activated receptor gamma knockout mice exhibit the spontaneous osteoarthritis phenotype. Am J Pathol 182: 1099-1106.
-
Hyde G, Boot-Handford RP, Wallis GA (2008) Col2a1 lineage tracing reveals that the meniscus of the knee joint has a complex cellular origin. J Anat 213: 531-538.
-
Hyde G, Dover S, Aszodi A, Wallis GA, Boot-Handford, RP (2007) Lineage tracing using matrilin-1 gene expression reveals that articular chondrocytes exist as the joint interzone forms. Dev Biol 304: 825-833.
-
Kague E, Gallagher M, Burke S, Parsons M, Franz-Odendaal T, et al. (2012) Skeletogenic fate of zebrafish cranial and trunk neural crest. PLoS One 7: e47394.
-
Park J, Gebhardt M, Golovchenko S, Perez-Branguli F, Hattori T, et al. (2015) Dual pathways to endochondral osteoblasts: a novel chondrocyte-derived osteoprogenitor cell identified in hypertrophic cartilage. Biol Open 4: 608-621.
-
Schwartz AG, Long F, Thomopoulos S (2015) Enthesis fibrocartilage cells originate from a population of Hedgehog-responsive cells modulated by the loading environment. Development 142: 196-206.
-
Lounev VY, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment AD, et al. (2009) Identification of progenitor cells that contribute to heterotopic skeletogenesis. J Bone Joint Surg Am 91: 652-663.
-
Gaj T, Gersbach CA, Barbas CF 3rd (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31: 397-405.
-
Buchan JG, Gray RS, Gansner JM, Alvarado DM, Burgert L, et al. (2014) Kinesin family member 6 (kif6) is necessary for spine development in zebrafish. Dev Dyn 243: 1646-1657.
-
Ablain J, Durand EM, Yang S, Zhou Y, Zon LI (2015) A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Dev Cell 32: 756-764.
-
Mandegar MA, Huebsch N, Frolov EB, Shin E, Truong A, et al. (2016) CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 18: 541-553.
-
Mendell JR, Rodino-Klapac LR (2016) Duchenne muscular dystrophy: CRISPR/Cas9 treatment. Cell Res 26: 513-514.
-
Sander JD, Joung JK (2014) CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32: 347-355.
-
Xu L, Park KH, Zhao L, Xu J, El Refaey M, et al. (2016) CRISPR-mediated Genome Editing Restores Dystrophin Expression and Function in mdx Mice. Mol Ther 24: 564-569.
-
Meyer MB, Benkusky NA, Pike JW (2015) Selective Distal Enhancer Control of the Mmp13 Gene Identified through Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) Genomic Deletions. J Biol Chem 290: 11093-11107.
-
Yang M, Zhang L, Stevens J, Gibson G (2014) CRISPR/Cas9 mediated generation of stable chondrocyte cell lines with targeted gene knockouts; analysis of an aggrecan knockout cell line. Bone 69: 118-125.
-
Willems B, Tao S, Yu T, Huysseune A, Witten PE, et al. (2015) The Wnt Co-Receptor Lrp5 Is Required for Cranial Neural Crest Cell Migration in Zebrafish. PLoS One 10: e0131768.
-
DeLaurier A, Eames BF, Blanco-Sánchez B, Peng G, He X, et al. (2010) Zebrafish sp7:EGFP: a transgenic for studying otic vesicle formation, skeletogenesis, and bone regeneration. Genesis 48: 505-511.
-
Renn J, Winkler C (2009) Osterix-mCherry transgenic medaka for in vivo imaging of bone formation. Dev Dyn 238: 241-248.
-
Spoorendonk KM, Peterson-Maduro J, Renn J, Trowe T, Kranenbarg S, et al. (2008) Retinoic acid and Cyp26b1 are critical regulators of osteogenesis in the axial skeleton. Development 135: 3765-3774.
-
Knopf F, Hammond C, Chekuru A, Kurth T, Hans S, et al. (2011) Bone regenerates via dedifferentiation of osteoblasts in the zebrafish fin. Dev Cell 20: 713-724.
-
Vanoevelen J, Janssens A, Huitema LF, Hammond CL, Metz JR, et al. (2011) Trpv5/6 is vital for epithelial calcium uptake and bone formation. FASEB J 25: 3197-3207.
-
Hammond CL, Schulte-Merker S (2009) Two populations of endochondral osteoblasts with differential sensitivity to Hedgehog signalling. Development 136: 3991-4000.
-
Mitchell RE, Huitema LF, Skinner RE, Brunt LH, Severn C, et al. (2013) New tools for studying osteoarthritis genetics in zebrafish. Osteoarthritis Cartilage 21: 269-278.
-
Hammond CL, Moro E (2012) Using transgenic reporters to visualize bone and cartilage signaling during development in vivo. Front Endocrinol (Lausanne) 3: 91.
-
Bowman TV, Zon LI (2010) Swimming into the future of drug discovery: in vivo chemical screens in zebrafish. ACS Chem Biol 5: 159-161.
-
Kaufman CK, White RM, Zon L (2009) Chemical genetic screening in the zebrafish embryo. Nat Protoc 4: 1422-1432.
-
de Vrieze E, Zethof J, Schulte-Merker S, Flik G, Metz JR (2015) Identification of novel osteogenic compounds by an ex-vivo sp7:luciferase zebrafish scale assay. Bone 74: 106-113.
-
Rosendahl K, Dezateux C, Fosse KR, Aase H, Aukland SM, et al. (2010) Immediate treatment versus sonographic surveillance for mild hip dysplasia in newborns. Pediatrics 125: e9-16.
-
Lowry RB, Sibbald B, Bedard T, Hall JG (2010) Prevalence of multiple congenital contractures including arthrogryposis multiplex congenita in Alberta, Canada, and a strategy for classification and coding. Birth Defects Res A Clin Mol Teratol 88: 1057-1061.
-
Bayat A, Petersen A, Møller M, Andersen G, Ebbesen F (2009) Incidence of fetal akinesia-hypokinesia deformation sequence: a population-based study. Acta Paediatr 98: 3-4.
-
Nayak SS, Kadavigere R, Mathew M, Kumar P, Hall JG, et al. (2014) Fetal akinesia deformation sequence: expanding the phenotypic spectrum. Am J Med Genet A 164A: 2643-2648.
-
Nowlan NC, Sharpe J, Roddy KA, Prendergast PJ, Murphy P (2010) Mechanobiology of embryonic skeletal development: Insights from animal models. Birth Defects Res C Embryo Today 90: 203-213.
-
Rolfe R, Roddy K, Murphy P (2013) Mechanical regulation of skeletal development. Curr Osteoporos Rep 11: 107-116.
-
Jahan E, Matsumoto A, Rafiq AM, Hashimoto R, Inoue T, et al. (2014) Fetal jaw movement affects Ihh signaling in mandibular condylar cartilage development: the possible role of Ihh as mechanotransduction mediator. Arch Oral Biol 59: 1108-1118.
-
Kavanagh E, Church VL, Osborne AC, Lamb KJ, Archer CW, et al. (2006) Differential regulation of GDF-5 and FGF-2/4 by immobilisation in ovo exposes distinct roles in joint formation. Dev Dyn 235: 826-834.
-
Roddy KA, Kelly GM, van Es MH, Murphy P, Prendergast PJ (2011). Dynamic patterns of mechanical stimulation co-localise with growth and cell proliferation during morphogenesis in the avian embryonic knee joint. J Biomech 44: 143-149.
-
Rolfe RA, Nowlan NC, Kenny EM, Cormican P, Morris DW, et al. (2014) Identification of mechanosensitive genes during skeletal development: alteration of genes associated with cytoskeletal rearrangement and cell signalling pathways. BMC genomics 15: 48.
-
Kahn J, Shwartz Y, Blitz E, Krief S, Sharir A, et al. (2009) Muscle contraction is necessary to maintain joint progenitor cell fate. Dev Cell 16: 734-743.
-
Lories RJ, Corr M, Lane NE (2013) To Wnt or not to Wnt: the bone and joint health dilemma. Nat Rev Rheumatol 9: 328-339.
-
Rayfield EJ (2005) Using finite-element analysis to investigate suture morphology: a case study using large carnivorous dinosaurs. Anat Rec A Discov Mol Cell Evol Biol 283: 349-365.
-
Nowlan NC, Murphy P, Prendergast PJ (2008) A dynamic pattern of mechanical stimulation promotes ossification in avian embryonic long bones. J Biomech 41: 249-258.
-
Brunt LH, Norton JL, Bright JA, Rayfield EJ, Hammond CL (2015) Finite element modelling predicts changes in joint shape and cell behaviour due to loss of muscle strain in jaw development. J Biomech 48: 3112-3122.
-
Roddy KA, Prendergast PJ, Murphy P (2011) Mechanical influences on morphogenesis of the knee joint revealed through morphological, molecular and computational analysis of immobilised embryos. PloS one 6: e17526.
-
Nowlan NC, Prendergast PJ, Murphy P (2008) Identification of mechanosensitive genes during embryonic bone formation. PLoS Comput Biol 4: e1000250.
