

Journal of Rheumatic Diseases and Treatment
A Pediatric Case of NMOSD with Positive Seroconversion of AQP4-ab after 5 Years and Subsequent Diagnosis of SLE
Aisha Bushra, Osman Farooq, Svetlana P Eckert, Caila B Vaughn, Rabheh Abdul Aziz and Bianca Weinstock-Guttman*
Department of Neurology, State University of New York at Buffalo, USA
*Corresponding author: Bianca Weinstock-Guttman, MD, Professor, Department of Neurology, State University of New York at Buffalo; Director, Jacobs MS Center for Treatment and Research, USA, E-mail: bweinstock-guttman@kaleidahealth.org
J Rheum Dis Treat, JRDT-3-051, (Volume 3, Issue 2), Case Report; ISSN: 2469-5726
Received: November 16, 2016 | Accepted: March 16, 2017 | Published: March 18, 2017
Citation: Bushra A, Farooq O, Eckert SP, Vaughn CB, Aziz RA, et al. (2017) A Pediatric Case of NMOSD with Positive Seroconversion of AQP4-ab after 5 Years and Subsequent Diagnosis of SLE. J Rheum Dis Treat 3:051. 10.23937/2469-5726/1510051
Copyright: © 2017 Bushra A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Neuromyelitis optica spectrum disorder (NMOSD) is a demyelinating disorder of the central nervous system that is distinct from multiple sclerosis and is primarily characterized by recurrent episodes of optic neuritis (ON) and/or longitudinal extensive transverse myelitis (LETM). The presence of aquaporin antibodies (AQP4-ab) in the serum and/or cerebrospinal fluid (CSF) is a highly specific biomarker for diagnosis, although repeated testing may be necessary to confirm its presence. Here, we report on a case of a 15-year-old African American female diagnosed with NMOSD following two recurrent brainstem events, which consistently tested seronegative for AQP4-ab for five years and maintained a stable clinical and magnetic resonance imaging (MRI) status. The patient was treated with mycophenolate mofetil (MMF) therapy for five years and remained without relapse by MRI. At that point therapy was discontinued. Repeat surveillance testing six months after discontinuation revealed high positive titers of AQP4-ab, despite the patient being asymptomatic. Patient ultimately presented with altered mental status, and subcutaneous nodules were noted, prompting a rheumatological evaluation, which was consistent with a diagnosis of systemic lupus erythematosus (SLE). Intense immunosuppression therapy provided full clinical and MRI recovery. Our case outlines the importance of monitoring AQP-4 antibody seropositivity in patients with a high clinical suspicion, despite initial negative tests results. This case also challenges the often-provided recommendation for therapy interruption after 5 years of clinical remission and demonstrates the possibility of development of other autoimmune disorders like SLE in conjunction with NMOSD.
Introduction
Neuromyelitis optica (NMO, Devic's Disease) is an inflammatory demyelinating disorder of the central nervous system (CNS) that preferentially damages the optic nerve and spinal cord [1]. The presence of antibodies to aquaporin-4 (AQP4-ab) supports the diagnosis of NMO and facilitates the differentiation of NMO from other inflammatory CNS pathologies, primarily multiple sclerosis (MS) [2]. The newly recommended definition known as NMO spectrum disorder (NMOSD) introduced the area postrema, brainstem and diencephalon as additional possible injury areas. The presence of aquaporin antibodies against water AQP-4 channels in the serum and/or CSF is a highly specific biomarker for the NMOSD diagnosis, although repeated testing is often necessary to confirm that the antibodies are present [3]. Here we report on a case of pediatric NMOSD, who was negative for AQP4-ab for 5 years while being treated with immunosuppressive (IS) therapy, but who converted to positive status following the discontinuation of IS therapy, and who later developed CNS lupus.
Case Report
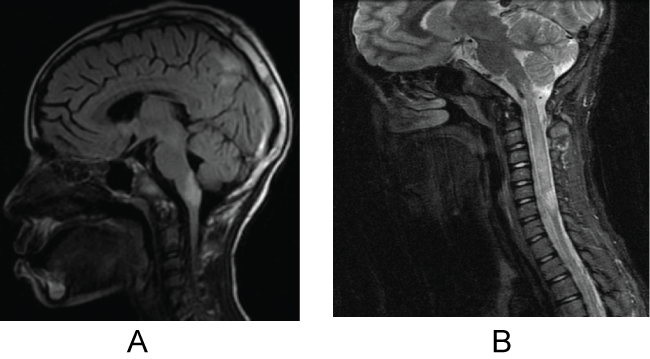
The patient is currently a 15-year-old African American female who initially presented with an acute episode of dysphagia and severe weight loss at age 8. On physical examination, she was found to have marked bulbar dysfunction with dysphagia and dysphonia, difficulty chewing, and subsequent weight loss with recurrent pulmonary aspiration and pneumonia. Please refer to Table 1 for a summary of patient's clinical course and treatments. A large brainstem lesion was noted on MRI (Figure 1a). A follow-up MRI performed five days later showed enlargement of the brainstem lesion as well as multiple areas of hyper-intensities and a diagnosis of acute disseminated encephalomyelitis (ADEM) was considered. Analysis of the cerebrospinal fluid (CSF) revealed no oligoclonal bands (OB's), glucose was 74 mg/dl, protein was 19 mg/dl, two nucleated cells, and no RBCs. After treatment with steroids and intravenous immunoglobulins (IVIG) her condition improved and she was again able to eat independently. Five months later, she presented with gait difficulties, repeated falls and distal paresthesias. The CSF analysis revealed no red blood cells, seven nucleated cells with 92% lymphocytes, glucose of 45 mg/dl and protein of 26 mg/dl, and was negative for OB's. AQP4-ab in the serum was negative, but her serum angiotensin converting enzyme (ACE) level was 70 units (Normal: 40 units). A brain MRI showed improvement in the previous brainstem lesion however, it also demonstrated a new lesion extending from C2 to C7 (Figure 1b). Her spinal cord lesion was suggestive of NMO given the extent of the lesion beyond three vertebral columns. A diagnosis of sarcoidosis was also considered due to the elevated ACE level. However, ACE in CSF was normal (< 1 unit/L), calcium in the urine was normal, and no lymphadenopathy was reported on the chest CT scan and, therefore, sarcoidosis was ruled out. The patient was treated with steroids, fully recovered and was started on preventive mycophenolate mofetil (MMF). On follow-up MRI three months later the brainstem lesion was nearly resolved and the cervical cord lesion showed significant improvement. The patient did not have optic neuritis at any time during her diagnostic work-up. Given the significant improvement on the images and normal CSF cytology, central nervous system malignancy was excluded. She continued on MMF for 4 years on 1500 mg/day and experienced improvement in her symptoms; her MRIs (brain and cervical) were completely normal over these 4 years. The following year a slow taper of MMF was implemented until the therapy was fully discontinued. Repeat testing of AQP4-ab during the acute as well as stable stages was consistently negative. Six months following the MMF discontinuation, while the patient was symptom free and with normal MRIs, a repeat NMO IgG test showed high positive levels (123 units/ml and a repeated test 1 month later was 140 units/ml). The patient was restarted on MMF on a slower titration. Two months later, now at age 14, she complained of increased generalized pain and visual problems. The repeat MRIs on presentation to the hospital were stable but the visual evoked potential (VEP) suggested prolonged conduction on the left side. An inpatient course of IV steroids helped her recover from the new symptoms, nearly back to baseline. A repeat lumbar puncture (LP) did not reveal oligoclonal bands, but did show IgG index of 0.3 (0.33-0.63). Further clinical course is also summarized in table 1. The patient then presented a month later with paresthesias in her left lower extremity (LLE) and again with vague complaints of blurry vision. Although no evidence of optic neuritis was found on funduscopic exam or MRIs, the patient again was treated with a 3-day course of IV steroids (methylprednisolone 1 g IV daily). She demonstrated clinical improvement along with new mild pachymeningeal enhancement seen on MRI that at that time was thought to be due to recent LP. A repeat LP was done, which showed again IgG 5.9 mg/dl with no OB's. At this time patient's MMF dose was increased to 1000 mg two times per day. In addition, at this presentation some nonspecific subcutaneous nodules were noted and as part of a vasculitis work up anti-nuclear antibodies (ANA) were tested, which on subsequent admission were noted to be highly positive (ANA titer of 1:2560).
.
Figure 1a : May 2010: Sagittal FLAIR revealed enlargement of the brainstem lesion as well as the presence of hemispheric lesions.
Figure 1b : October 2010: Sagittal T2 MRI of the C-spine revealed a non-enhancing lesion extending from C2 to C7.
View Figure 1
![]()
Table 1: Timeline of events.
View Table 1
Five days after discharge from the hospital, the patient had deterioration in her affect, and became progressively withdrawn from her family and stopped communicating basic needs. She stopped talking or getting out of bed, required assistance with feeding and activities of daily living, became bladder and bowel incontinent and returned to the hospital. On physical exam at that time, she did not follow any commands, had midline gaze without tracking, had diffusely increased tone, and generalized swelling, with persistent non-specific subcutaneous skin nodules and persistent variable tachycardia in the 140's-160's, with occasional fever up to 38 degrees Celsius.
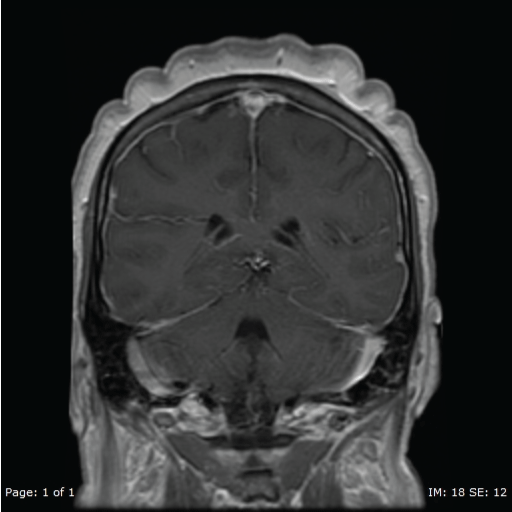
A repeat MRI of the brain showed diffuse prominent pachymeningeal enhancement (Figure 2), excessive for what otherwise would be expected with a LP performed 4 weeks prior. Another repeat LP was done and was negative for infectious etiologies. At this point she had positive serum AQP4-ab of 123 IU/ml, elevated thyroid microsomal antibodies (while remaining thyroid function tests were normal) and the ANA titer from previous admission returned positive at 1:2560. An autoimmune encephalitis, possibly SLE encephalitis, was presumed.
.
Figure 2: Repeat MRI November 2015. MRI T1 with contrast showing diffuse pachymeningeal enhancement.
View Figure 2
Because of the development of pachymeningeal enhancement on MRI, rheumatology evaluated patient as well and a complete lupus work-up was performed for possible CSN lupus. She was diagnosed with neuropsychiatric SLE based on Systemic Lupus International Collaborating Clinics Classification Criteria for Systemic Lupus Erythematosus [4]. Her criteria included: arthritis, positive ANA (1:2560), positive ENA smith, low C3 and low C4, and neurologic finding with acute confusional state in the absence of other causes, including toxic, metabolic, uremia, and drugs. In addition, her ENA SSA was positive as well as ENA RNP, IGG was significantly elevated (6670 mg/dl), and subcutaneous nodules biopsy showed panniculitis; that all can be seen in lupus, but are not considered to be strict criteria. The patient was treated with plasmapheresis, but demonstrated no improvement and completed only 5 out of a 6-day course due to transient bacteremia that required a central catheter change. At this time patient had persistently increased tone with some thrashing movements on the bed, irregular head movements and tremulousness with some concern for seizures. Long term video EEG monitoring was performed and showed evidence of diffuse encephalopathy only, without any evidence of seizure activity. There was some concern that the patient's symptoms might have been related to an atypical NMO presentation, and patient then received a starting dose of an IV rituximab infusion for treatment after she demonstrated no response to IV steroid therapy and five days of plasma exchange (PLEX). At the same time, patient was continued on high dose MMF. The patient continued to be persistently tachycardic and unresponsive.
By approximately the fifth week of admission a trial of a 5-day course of IVIG, 0.4 mg/kg/day was started, and the patient began to improve, although very slowly. Initially on physical exam her tone improved. Her generalized body edema and subcutaneous nodules subsequently resolved as well. The patient recovered significantly over the next few weeks, though she required intensive physical therapy initially, and then went back to school after 4 months, nearly at baseline functioning. Her repeat MRI became normal while continuing on a high dose (3,000 mg) of MMF, monthly steroids and IVIG. In addition, our patient had her menarche at 12 years of age and had regular periods until she had her first acute exacerbation after 5 years of her disease. Since then she has been in amenorrhea. She denies any blurry vision, galactorrhea, extra body hair or acne.
Discussion
NMO has variable clinical manifestations that are not limited to the characteristic features of optic neuritis and transverse myelitis. Brain abnormalities in NMO and NMOSD are relatively common, and patients may present with brain involvement at disease onset without any additional symptoms [5]. Therefore, NMO may be difficult to distinguish from ADEM and MS, particularly in the pediatric population. The new diagnostic criteria of NMOSD include at least one of the six clinical characteristics (optic neuritis, transverse myelitis, area postrema syndrome, acute brain stem syndrome, symptomatic narcolepsy and symptomatic cerebral syndrome) along with detection of AQP4-ab, MRI findings are usually helpful in localizing the extent of pathology [6]. Our patient's MRI findings were suggestive of NMOSD during her relapses, but were fully recovered within 1 year on MMF therapy.
NMO IgG antibodies are identified during active NMOSD disease and often they persist although fluctuations with decreased levels are seen during effective treatment [7]. A stable disease under effective immunosuppressive therapy often may convert the AQP4-ab to a negative status [3]. Repeated AQP4-ab testing should be considered as a useful biological marker for monitoring NMOSD in individual patients and/or response to therapy. Prior studies indicate that AQP4-ab antibodies may be detected in greater than 75% of patients with NMO, and that the clinical course of NMO varies between AQP4-ab positive and negative patients [8]. The sensitivity of assays available for antibody detection varies by 20%, which may contribute to a false negative test result in patients with NMO [1]. One study showed the sensitivity of AQP4 antibody assay was 91% for NMO and 85% for high-risk syndrome, but the specificity is 100% for NMOSD [9]. Repeated testing may be required, as NMO antibodies either in CSF or serum may become positive 3 years after the onset symptoms [10].
The presence of AQP4-ab at the time of an initial event suggestive of NMOSD is associated with high risk for relapses. Therefore, IS therapy is typically implemented after an initial event, especially in the presence of AQP4-ab. If patient fails to respond to corticosteroid therapy, plasma exchange (PLEX) comes into consideration and for a long-term decrease in relapses, rituximab can be effective. The value of therapies in acutely deteriorating patients can be inspected prospectively and should be distinguished from efficacy of long-term therapies intended at the prevention of relapses [11]. The often-used recommendation to consider discontinuing the therapy after 5 years of stable disease has to be carefully considered as the new 2015 Weinshenker reviews are changed [6]. Our patient developed high titer of ANA, anti ENA, anti ENA SSA, ENA RNP, and ENA SSA antibodies with low C3 with arthritis and subcutaneous nodules, and was subsequently diagnosed with SLE with CNS involvement, in addition to AQP4-ab positive status. The neurologic course could be an independent process if clinically evident SLE, Sjogren's, or positive autoantibodies coexist with NMO, or it may be a complication of a systemic disease making it difficult to make the accurate diagnosis which can ultimately also affect the choice of treatment in the acute setting and maintenance immunosuppressive therapy [12].
An NMOSD diagnosis results in a higher probability of developing other autoimmune diseases, including myasthenia gravis, inflammatory bowel disease, SLE, rheumatoid arthritis, Sjogren's syndrome and immune thrombocytopenic purpura [13]. SLE associated with LETM disease modifying agents and NSAIDs are part of the treatment [14]. In a previous case series, use of immunosuppressants with corticosteroids has shown improvement, and in a separate study, a combination of cyclophosphamide with corticosteroids (CTX) resulted in 14% complete resolution of the symptoms and 59% partial recovery [15]. Rituximab appears to show disease stabilization and decrease in relapses in patients of NMO, [16] where interferon beta did not show any efficacy in preventing relapses and even resulted in exacerbation of disease [17]. NMOSD patients require immunosuppressants to decrease the chances of relapses. Either azathioprine with prednisone or rituximab is recommended as a first line prophylactic therapy in NMO by EFNS guidelines while MMF (1-3 g/day), mitoxantrone (12 mg/m2) every 3 months or cyclophosphamide (7-25 mg/kg monthly for 6 months) are potential second line therapies [18].
Our report highlights the importance of re-testing patients for NMO-IgG antibodies even with normalized MRI's, especially if discontinuation of therapy is being considered. This is particularly important in the pediatric population in which the immune system may change along with hormonal and sexual development [19]. We additionally recommend that retesting for AQP-4 ab should be performed even after discontinuation of treatment, as a monitoring tool of disease activity. This case should also emphasize the importance of the co-existence of AQP4-ab with other autoimmune disorders, like SLE and Sjogren's syndrome [20].
References
-
Jarius S, Wildemann B (2013) Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol 23: 661-683.
-
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG (2006) Revised diagnostic criteria for neuromyelitis optica. Neurology 66: 1485-1489.
-
Weinstock-Guttman B, Miller C, Yeh E, Stosic M, Umhauer M, et al. (2008) Neuromyelitis optica immunoglobulins as a marker of disease activity and response to therapy in patients with neuromyelitis optica. Mult Scler 14: 1061-1067.
-
Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, et al. (2012) Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 64: 2677-2686.
-
Kim W, Kim SH, Huh SY, Kim HJ (2012) Brain abnormalities in neuromyelitis optica spectrum disorder. Mult Scler Int.
-
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, et al. (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85: 177-189.
-
Nakatsuji Y (2014) Blood and cerebrospinal fluid biomarkers in MS/NMO. Nihon Rinsho 72: 1970-1975.
-
Waters P, Jarius S, Littleton E, Leite MI, Jacob S, et al. (2008) Aquaporin-4 antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis. Arch Neurol 65: 913-919.
-
Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, et al. (2007) Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 130: 1235-1243.
-
Chitnis T, Ness J, Krupp L, Waubant E, Hunt T, et al. (2016) Clinical features of neuromyelitis optica in children: US Network of Pediatric MS Centers report. Neurology 86: 245-252.
-
Scott TF, Frohman EM, De Seze J, Gronseth GS, Weinshenker BG, et al. (2011) Evidence-based guideline: clinical evaluation and treatment of transverse myelitis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 77: 2128-2134.
-
Karim S, Majithia V (2009) Devic's syndrome as initial presentation of systemic lupus erythematosus. Am J Med Sci 338: 245-247.
-
Iyer A1, Elsone L, Appleton R, Jacob A (2014) A review of the current literature and a guide to the early diagnosis of autoimmune disorders associated with neuromyelitis optica. Autoimmunity 47: 154-161.
-
Nardone R, Fitzgerald RT, Bailey A, Zuccoli G (2015) Longitudinally extensive transverse myelitis in systemic lupus erythematosus: case report and review of the literature. Clin Neurol Neurosurg 129: 57-61.
-
Espinosa G1, Mendizábal A, Mínguez S, Ramo-Tello C, Capellades J, et al. (2010) Transverse myelitis affecting more than 4 spinal segments associated with systemic lupus erythematosus: clinical, immunological, and radiological characteristics of 22 patients. Semin Arthritis Rheum 39: 246-256.
-
Jacob A, Weinshenker BG, Violich I, McLinskey N, Krupp L, et al. (2008) Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol 65: 1443-1448.
-
Tanaka M, Tanaka K, Komori M (2009) Interferon-beta(1b) treatment in neuromyelitis optica. Eur Neurol 62: 167-170.
-
Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, et al. (2010) EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol 17: 1019-1032.
-
McMurray RW, May W (2003) Sex hormones and systemic lupus erythematosus: review and meta-analysis. Arthritis Rheum 48: 2100-2110.
-
Amorim AL, Cabral NC, Osaku FM, Len CA, Oliveira EM, et al. (2016) Association between demyelinating disease and autoimmune rheumatic disease in a pediatric population. Rev Bras Reumatol.
