

Journal of Rheumatic Diseases and Treatment
Osteoimmunological Aspects on Inflammation and Bone Metabolism
Uwe Lange*, Gabriel Dischereit, Elena Neumann, Klaus Frommer, Ingo H. Tarner and Ulf Müller-Ladner Kerckhoff-Klinik
Department of Rheumatology, Osteology and Physical Medicine, University of Giessen, Germany
*Corresponding author: Professor Uwe Lange, Department of Rheumatology, Osteology and Physical Medicine, University of Giessen, Benekestr. 2-8, D- 61231 Bad Nauheim, Germany, Tel: 0049(0)6032-996-2101, Fax: 0049(0)6032-996-2185, E-mail: U.Lange@kerckhoff-klinik.de
J Rheum Dis Treat, JRDT-1-008, (Volume 1, Issue 2), Review Article; ISSN: 2469-5726
Received: February 12, 2015 | Accepted: April 07, 2015 | Published: April 09, 2015
Citation: Lange U, Dischereit G, Neumann E, Frommer K, Tarner IH, et al. (2015) Osteoimmunological Aspects on Inflammation and Bone Metabolism. J Rheum Dis Treat 1:008. 10.23937/2469-5726/1510008
Copyright: © 2015 Lange U, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Bone remodelling is characterized by a balance between bone resorption and bone formation. The osteoblasts are responsible for bone synthesis and the osteoclasts for bone resorption. A finely adjusted interaction between molecular mechanisms results, via cytokines, hormones and growth factors, in homeostasis of bone metabolism. Here, the RANK/RANKL/OPG-system is actively involved in the differentiation and function of osteoclasts and is known to play a central role in the majority of pathophysiological mechanisms. Also the Wnt and BMP signalling pathways play a major role in osteoblast differentiation and bone remodeling. An increased osteoclast activity contributes to inflammatory and destructive osteocatabolic manifestations and/or osteoporosis whereas an increased osteoblast activity can result in osteopetrosis. This overview describes the known relevant pathophysiological metabolic pathways in this remodelling process, especially the effect of inflammation on bone metabolism, and outlines the links from bench to bedside.
Keywords
Bone metabolism, Remodelling, Osteoporosis, RANK/RANKL/OPG, Wnt, DKK1, BMP
Introduction
The bone is subject to constant remodeling. This enables bones to adapt to changing strain or stress situations and required reparative mechanisms in cases such as microcracks or fragility fractures which hazards the optimal bone structure. Thus, bone reconstruction in healthy individuals is dependent on a balanced relationship between bone formation and bone loss [1,2]. This balance is also essential for the process of achieving an optimal peak bone mass which takes place before adulthood. At the cellular level, the remodeling process is controlled through the balanced interaction between osteoblasts and osteoclasts whereby the cellular balance of bone metabolism is safeguarded through molecular mechanisms (e.g. cytokines, hormones, and growth factors). In addition, factors such as age, gender, post-menopausal estrogen loss, physical activity, and various drugs also influence bone metabolism [2,3].
Over the past years, molecular biologic studies have significantly broadened our scope of knowledge regarding the regulation of bone metabolism. In particular, the immune system and bone metabolism are so closely intertwined that pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1, IL-6, and IL-17 could be identified as stimulators in the formation of osteoclasts. They are therefore essential mediators in bone resorption, primarily in chronic inflammatory diseases [4,5].
The basic requirement for the maintenance of bone homeostasis is a sufficient distinction between osteoclasts and osteoblasts. Two cytokines, namely the macrophage colony stimulating factor (M-CSF) and the receptor activator of NF-κB ligand (RANKL), are of significant importance in osteoclastogenesis which, as solitary cells, can deteriorate or destroy bones. The osteoclasts themselves originate from monocytic precursor cells. If one of these messengers is missing, the formation of osteoclasts comes to a halt resulting in increased bone formation, i.e. osteopetrosis [2,6,7]. In addition to the proinflammatory factors TNF-α and IL-6, the following factors play a significant role in osteoblast differentiation from mesenchymal stem cells:
� The Wnt / wingless protein family
� Bone Morphogenetic Protein (BMP)
� Transforming Growth Factor beta (TGF-β)
� Fibroblast Growth Factor (FGF)
� Insulin-like Growth Factor (IGF)
A �reverse� disorder in the osteoblastic system results in osteoporosis [2,6,7], which is characterized by low bone density and micro-architectural alterations in bones. Further characteristics are successively decreased bone stability, increased fracture susceptibility and / or inflammatory destructive manifestations.
The Molecular Mechanisms of Inflammatory Mediators
Secondary osteoporosis is a frequent complication in various inflammatory rheumatic diseases, usually combined with increased levels of pro-inflammatory cytokines [2,5,7]. In this situation bone mass loss can be extremely high in the first three to six (sometimes twelve) months with the use of glucocorticoid medication (up to 20% main bone mass loss [�very high turnover� respectively �fast looser�]) [8], and the use of anti-rheumatic medications such as NSAID and DMARD can add to this alteration in bone metabolism.
The pathogenetic aspects of bone mass loss in rheumatoid diseases includes bone-related mechanisms (i.e. inflammatory activity, inactivity, visceral association), effects of therapy, and factors unrelated to the underlying disease (e.g. post-menopause, pre-existing bone density loss, and beneficial effects of other secondary illnesses).
Inflammation, the RANK/RANKL/OPG, Wnt and BMP Signaling Pathways and Bone Metabolism
The influence of pro-inflammatory cytokines on bone metabolism in the form of bone mass loss is especially evident with the occurrence of inflammation since TNF-α, IL-1, IL-6 and IL-17 contribute to increased osteoclastogenesis [5,9-12]. Thus, a connection between the extent of the inflammatory reaction and bone metabolism manifests itself in enhancing fracture susceptibility: patients with a C-reactive protein (CRP) of >3mg/l have a fracture risk eight times greater than persons of similar age, gender, health, with a CRP < 1mg/l. An inverse relationship between inflammation and bone formation and bone loss markers is also evident in a CRP range that is < 7.5mg/l. On the contrary, a direct correlation between CRP and the biochemical markers for bone resorption could be identified in cases of increased inflammation (CRP >7.5mg/l) [5], indicating that higher levels of cytokines augment osteoclastogenesis and bone resorption.
Osteoclast-mediated bone resorption is controlled in particular by the key regulators RANKL, its receptor RANK (receptor activator of nuclear factor-κB) and by osteoprotegerin (OPG). The RANK/RANKL/OPG system plays a prominent role in bone resorption [7,12-14] (Figure 1). Both osteoblasts and T-lymphocytes activated by inflammatory conditions as well as B-cells and fibroblasts express RANKL, which is required for the osteoclast differentiation from precursor cells. RANKL docks on its receptor RANK which is expressed by the precursor cells of osteoclasts and activates cellular signal transduction cascades after attaching. The signal transduction cascades ultimately activate the osteoclasts, thereby increasing osteoclastogenesis. The interaction between RANKL and RANK with mature osteoclasts further leads to their activation and a higher survival rate. In addition, osteoblasts express OPG which serves as a decoy receptor for RANKL. Conversely its effect can be neutralized by OPG as being a physiological regulator in bone homeostasis.
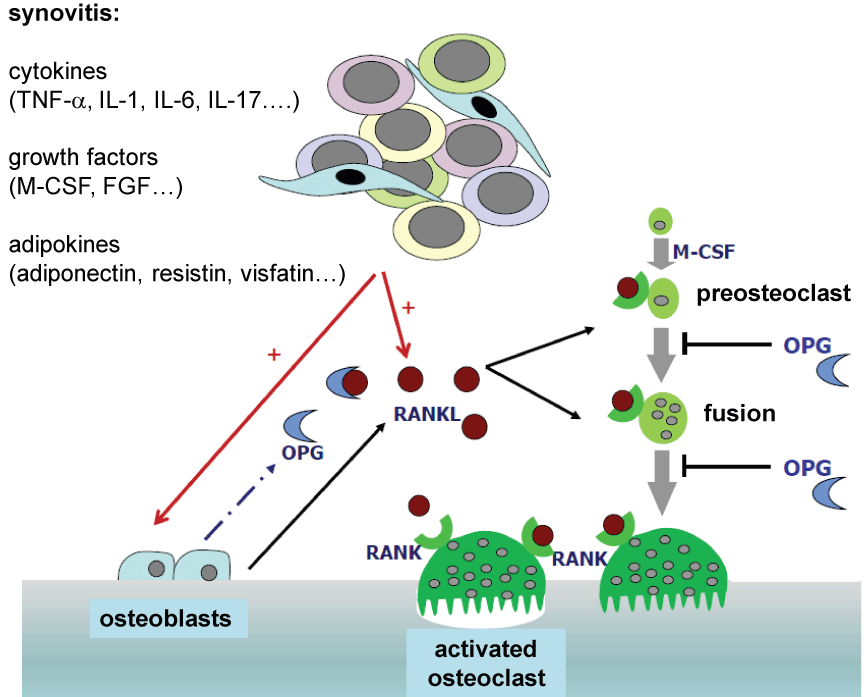
.
Figure 1: The central role of the RANK/RANKL/OPG-system in the bone remodeling process
TNF-α: Tumor Necrosis Factor α, IL-1: Interleukin-1, IL-6: Interleukin-6, IL-17: Interleukin-17, M-CSF: Macrophage Colony Stimulating Factor, FGF: Fibroblast
Growth Factor, OPG: Osteoprotegerin, RANK: Receptor Activator of NF-κB, RANKL: Receptor Activator of NF-κB Ligand
View Figure 1
Theoretically, an elevated ratio between RANKL and OPG results in bone loss; a balanced ratio or an inhibition in the RANK-response suppresses osteoclast activation and thereby the bone resorption as well [13].
The formation of osteoclasts through the induction of RANKL on mesenchymal cells and activated T-lymphocytes is increased by proinflammatory molecules such as TNF-α [10-12] and also directly facilitates osteoclastogenesis by means of attaching to the TNF- α-receptor-1 on the surface of monocytes.
The pro-inflammatory cytokine IL-1 also plays a significant role in bone metabolism. This cytokine induces the formation of RANKL and contribute to TNF-α-triggered bone resorption vice versa. Bone resorption induced by TNF-α is considerably limited in the absence of IL-1 by means of a diminished RANK expression leaving the osteoclast precursor cells resistant to stimulation by RANKL [2].
Likewise, IL-17 also plays a key role in bone metabolism as a connector between T-cells and bone resorption [2,3,15-17]. IL-17 is primarily expressed in Th-17 cells which also facilitate osteoclast formation while exhibiting pro-inflammatory characteristics. As inductor for the expression of TNF-α and IL-1, IL-17 is considered to be the main trigger in osteoclastogenesis. Furthermore, IL-17 induces the production of a number of other factors (e.g. IL-6, TNF-α, IL-1β, TGF-β, G-CSF, GM-CSF). In vivo experiments have shown that an inhibition of IL-17 leads to a suppression of joint destruction in collagen-induced arthritis [18,19], resulting in clinical trials evaluating the protective role of anti-IL-17 biologics. Another cytokine, IL-6, stimulates osteoclastogenesis by means of expression in RANKL. In particular, IL-6 plays an integral role in the osteo-destruction of multiple myeloma [20,21].
Additionally, bone remodeling relies upon two other important pathways that regulate osteoblast differentiation and function, the Wingless (Wnt) and bone morphogenetic protein (BMP) signaling pathways. Wnt signaling plays a key role in osteoblast differentiation and bone formation [22]. Various antagonists inhibit Wnt signaling and thus inhibit bone formation, including secreted frizzled-related proteins (sFRPs), the Dickkopf family members (DKKs), and sclerostin [23]. sFRPs induce a downregulation of the Wnt signaling by binding Wnt proteins. Overexpression of sFRP1 in osteoblasts suppresses Wnt signaling and induces apoptosis, whereas overexpression in mouse models has been shown to decrease bone density [24,25]. Furthermore, the deletion of the sFRP1 gene in mice results in increased trabecular bone mineral density [25]. Within the Dickkopf protein family, DKK1 inhibits bone formation by antagonizing Wnt signaling. An overexpression of DKK1 results in osteopenia in mice, whereas suppressed expression leads to high bone mass [26,27]. In a tumor necrosis factor (TNF) driven arthritis model it was shown that blockade of DKK1 induces fusion of the sacroiliac joints [28]. Also, inhibition of DKK1 transforms the destructive phenotype of some mouse models of arthritis into a remodeling phenotype in the peripheral joints [29].
Furthermore, an increase or decrease in serum levels of DKK1 has been associated with inflammatory diseases such as rheumatoid arthritis (RA) and ankylosing spondylitis (AS) [30,31]. Clinically, DKK1 serum levels are significantly increased in patients with RA compared to healthy controls. Additionally, TNF-α inhibitor therapy in RA patients has been shown to reduce elevated DKK1 levels to a near normal range [30]. In contrast, patients with AS presented DKK1 levels below the normal range as compared to healthy controls. Of interest, DKK1 is an inhibitor of Wnt signaling and thus an inhibitor of bone formation, whereas lower DKK1 levels would thus permit bone formation. Functional activity of DKK1 was observed in serum samples of AS patients [31]. Furthermore, patients with higher functional levels of DKK1 showed less syndesmophyte formation of the spine than patients with lower DKK1 activity. This suggests that lower levels or decreased activity of DKK1 may contribute to the excessive bone formation seen in AS, and that DKK1 may serve as a biomarker for osteocyte formation.
The BMP pathway plays also a key role in osteoblasts differentiation and bone remodeling. The most well-known BMP signaling cascade involves activation of Smad proteins. The binding of BMP ligands to their membrane-bound receptors results in phosphorylation of intracellular SMADS (SMADs 1 and 5), which associate with SMAD4 to form a complex that enters the nucleus to promote gene transcription [32]. It has been demonstrated that inhibition of BMPs can prevent peripheral joint ankylosis in mice [33]. To sum up, BMPs have been shown to play a role in inflammatory diseases, specifically in contributing to bone formation in AS [33]. Of note, proinflammatory cytokines can perturb the Wnt and BMP signaling pathways [34].
Anti-CCP Antibodies
An additional underlying mechanism been discovered with regard to the clinical observation that anti-CCP antibodies lead to augmented bone loss in rheumatoid arthritis. Here, anti-CCP antibodies which are in the context of inflammatory arthritides such as rheumatoid arthritis, present in over two-thirds of RA patients, can directly stimulate osteoclast formation. These antibodies are also considered prognostic parameters for a destructive process and therefore, it can be speculated that catabolic bone alterations in rheumatoid arthritis take place prior to over inflammatory destructive manifestations by means of this mechanism [35].
Adipokines
Additional potent mediators known for their immunomodulatory potential can act on bone underlying. Adipokines with immunomodulatory potential, such as adiponectin, resistin, and visfatin have been investigated with respect to their influence on pro-inflammatory and pro-destructive/fibrotic processes in rheumatoid diseases such as rheumatoid arthritis [36].
Adiponectin
The adipokine adiponectin exhibits predominantly immunomodulatory effects. In addition, it influences the function and the differentiation of central bone metabolism cells. For example, the globular isoform of adiponectin is a potential inhibitor of both TLR4-ligand-induced osteoclastogenesis and TNF/RANKL-induced osteoclast differentiation [37]. Adiponectin induces also RANKL and inhibits OPG expression in human osteoblasts. Suppression of AdipoR1, an adiponectin receptor, reverses adiponectin-induced effects with respect to RANKL and OPG expression [36,38,39]. Adipokines also have the ability to influence central bone metabolism cells. However at present it is still unclear how the (local) adipokine expression influences bone erosion or osteoporosis processes in detail in the context of rheumatoid diseases. If and how adiponectin has / holds therapeutic relevance for rheumatoid diseases is therefore currently being studied in various research groups.
Resistin
Similar to adiponectin, resistin inherits immunomodulatory potential. For example, resistin induces the secretion of cytokines such as TNF, IL-6, and IL-12 in inflammatory cells [36]. This led to the hypothesis that resistin, in contrast to adiponectin, which can function both as dependent on local conditions and as pro- and anti-inflammatory, is a pure pro-inflammatory factor. In the case of rheumatoid arthritis, serum levels of resistin are increased [36]. Increased levels of resistin in the synovium are also evident locally in rheumatoid arthritis, and, as with adiponectin, resistin is locally expressed in various disease-associated cells [36]. Resistin is, for example, induced in peripheral blood mononuclear cells (PBMC) by pro-inflammatory cytokines such as IL-6 and TNF. Conversely, resistin induces these factors in PBMC which leads to a pro-inflammatory feedback [36]. Resistin is also expressed in bone metabolism cells, for example in bone marrow stem cells and in mature human osteoblasts [40]. Recombined resistin can even increase the number of differentiated osteoclasts and the NFκB promoter activity during osteoclastogenesis in vitro [40].
Visfatin
Visfatin, also known as pre-B-cell colony-enhancing factor (PBEF), has been termed �visfatin� because of its expression in visceral fat tissue. Visfatin has the ability to bind the insulin receptor (IR) and thereby induce insulin-mimetic effects in various cells [36]. In addition, visfatin, or PBEF, is a pleiotropic protein with immunomodulatory potential and, like resistin, appears to experience predominantly pro-inflammatory effects. Visfatin / PBEF, induces cytokines such as IL-1, TNF, and IL-6 in monocytes. The concentration of visfatin / PBEF in synovial fluid and serum increases with rheumatoid arthritis [36]. Similar to adiponectin and resistin, visfatin / PBEF is locally expressed in synovium in a variety of cells [36,41]. In rheumatoid arthritis, the pro-inflammatory potential of visfatin / PBEF is reflected by diverse groups of various synovial effector cells [36,41]. Little is known about the influence of visfatin on bone metabolism. Nevertheless, it seems to wield insulin-similar effects on human osteoblasts. Insulin receptors are expressed in osteoblasts whereby visfatin / PBEF could induce the phosphorylation of IR and IR substrates [42]. Furthermore, the glucose intake, proliferation, and expression of collagen type 1 in human osteoblasts are induced whereas the osteocalcin secretion was reduced. Osteoclastic precursor cells, which are differentiated from PBMC, were suppressed by visfatin / PBEF from differentiating into mature, full-bodied osteoclasts [43]. This is also manifested in the suppression of osteoclast markers [43]. Thus, it appears that visfatin / PBEF, similar as resistin, influences the cells which are central to bone metabolism.
In conclusion, the etiology of bone density loss in inflammatory rheumatoid diseases is multifactorial. Pro-inflammatory cytokines act as stimulators of osteoclastogenesis and are thereby essential mediators in bone resorption. In this scenario, adipokines possess immunomodulatory potential and can influence cells central to bone metabolism.
From Bench to Bedside
A number of clinical studies concerning, i.e. rheumatoid arthritis and ankylosing spondylitis determine the fundamental role of TNF-α in the induction of osteoclasts and bone resorption. It could be shown that an adequate TNF-α inhibition is able to neutralize the inflammation-mediated alterations in bone metabolism, i.e. increased bone formation and reduced bone resorption, it also leads to a cessation or amelioration of radiographical erosive changes in patients with rheumatoid arthritis [44-48]. In this process, the relationship between RANKL and OPG shifts towards bone preservation in which OPG induces and RANKL decreases [45,48]. Furthermore, under the influence of TNF-β, this leads to the apoptosis of osteoclasts. The reduction of bone resorption due to a TNF-α inhibition may be restored through a reduced, direct stimulation of osteoblast formation and a reduced expression of RANKL in inflammation cells. The osteoprotective influence (bone neo-formation) of TNF-α could be recently be demonstrated by results revealing the interrelations between TNF-α and bone-inhibiting cytokines: TNF-α can induce Wnt-antagonists such as dickkopf (DKK-1) and thereby inhibit bone formation (Figure 2).
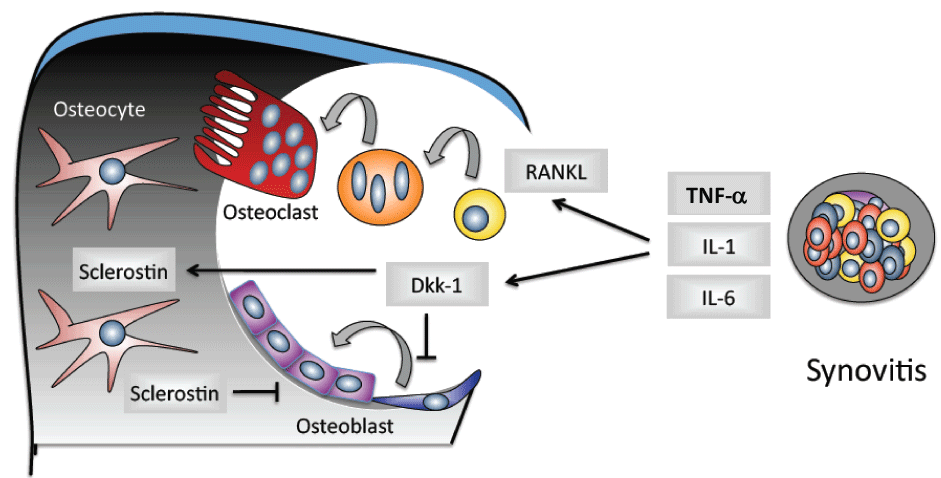
.
Figure 2: Inhibition of the bone formation by the Wnt-antagonist dickkopf (DKK-1) during the inflammation process.
View Figure 2
An inhibition of the radiographical progress in rheumatoid arthritis could also be shown when using different TNF-blockers [49-51] and the IL-6-receptor-blocker tocilizumab [52,53] even when no sufficient repression of the overall disease activity could be achieved [54,55]. In a one year prospective study, an osteoprotective effect on the bone metabolism and bone density could be confirmed by IL-6-Receptor inhibition in patients with active rheumatoid arthritis who had previously been treated with TNF-blockers [15].
With respect to active ankylosing spondylitis, it could be shown that the RANKL level and the RANKL/OPG ratio significantly increase with reduced bone density and radiographic signs of an existing inflammation [56]. In this context, the results of two current studies on serial radon-hyperthermia in the case of ankylosing spondylitis with a decrease in RANKL/OPG ratio are fascinating [57,58]. In these studies, the fall of osteo-catabolic and the rise of osteo-anabolic cytokines reflect the molecular basis for a decrease in osteoclastic bone loss in the context of an inflammation-conditioned secondary osteoporosis rather than through the influence of unaccompanied physical medicine. Among other aspects, it can be speculated that the systematic RANKL-secretion reduces T-cells and other osteocatabolic factors. Along this line, the observed decline of the TNF-α serum level could also be the result of a reduced expression of T-cells and other immune cells.
A therapy with the RANKL-inhibitor denosumab did not result in an improvement in arthritic flares in rheumatoid arthritis. The inhibition of osteoclasts, however, resulted in the progression of erosive bone alterations [59] and in an increase in peripheral and axial density [60]. A significant loss of the bone loss marker CTX-I could be shown in vitro [59]. Moreover an IL-17 blockade presents an additional attractive cytokine-based therapy option [61,62].
In conclusion, a therapeutic suppression of the inflammation process leads to a suppression of bone loss. In addition, physical therapy measures affect central messengers of the inflammation process as well as interactions between immune system cells and bone cells. As result various therapeutic options, which are currently being investigated are attractive future candidates in modulating the RANK/RANKL/OPG system.
Conclusion Parts
� Bone homeostasis is based on a finely balanced interaction among molecular mechanisms, cytokines, hormones, and growth factors.
� The effects of proinflammatory cytokines on osteoclasts and osteoblasts are numerous and are still being revealed.
� A close interrelation exists between the immune system and bone cells. Pro-inflammatory cytokines function as stimulators of osteoclastogenesis.
� The pathogenetic aspects of bone loss in inflammatory rheumatoid diseases focus on disease-associated mechanisms, inactivity, therapeutic results, and factors not to the underlying disease.
� A sufficient suppression of inflammatory symptoms is accompanied by a clinical improvement with a clear �osteoprotective� effect.
� Many components of the Wnt pathway are potential targets for the development of new drugs.
References
-
Hadjidakis DJ, Androulakis II (2006) Bone remodeling. Ann N Y Acad Sci 1092: 385-396.
-
Neumann E, Schett G (2007) Bone metabolism: molecular mechanisms. Z Rheumatol 66: 286-289.
-
Charatcharoenwitthaya N, Khosla S, Atkinson EJ, McCready LK, Riggs BL (2007) Effect of blockade of TNF-alpha and interleukin-1 action on bone resorption in early postmenopausal women. J Bone Miner Res 22: 724-729.
-
Kostenuik PJ (2005) Osteoprotegerin and RANKL regulate bone resorption, density, geometry and strength. Curr Opin Pharmacol 5: 618-625.
-
Schett G, Kiechl S, Weger S, Pederiva A, Mayr A, et al. (2006) High-sensitivity C-reactive protein and risk of nontraumatic fractures in the Bruneck study. Arch Intern Med 166: 2495-2501.
-
Cohen MM Jr (2006) The new bone biology: pathologic, molecular, and clinical correlates. Am J Med Genet A 140: 2646-2706.
-
Neumann E, Gay S, M�ller-Ladner U (2005) The RANK/RANKL/osteoprotegerin system in rheumatoid arthritis: new insights from animal models. Arthritis Rheum 52: 2960-2967.
-
Lange U, M�ller-Ladner U (2007) [Glucocorticoid induced osteoporosis]. Z Rheumatol 66: 129-136.
-
Goldring SR (2003) Inflammatory mediators as essential elements in bone remodeling. Calcif Tissue Int 73: 97-100.
-
Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, et al. (2000) TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest 106: 1481-1488.
-
Li P, Schwarz EM, O'Keefe RJ, Ma L, Boyce BF, et al. (2004) RANK signaling is not required for TNFalpha-mediated increase in CD11(hi) osteoclast precursors but is essential for mature osteoclast formation in TNFalpha-mediated inflammatory arthritis. J Bone Miner Res 19: 207-213.
-
Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM (2003) Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest 111: 821-831.
-
Hofbauer LC, Schoppet M (2004) Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. JAMA 292: 490-495.
-
Hofbauer LC (2006) Pathophysiology of RANK ligand (RANKL) and osteoprotegerin (OPG). Ann Endocrinol (Paris) 67: 139-141.
-
Wettich T, Dischereit G, M�ller-Ladner U, Lange U (2014) Effects of tocilizumab therapy on bone mineral density, markers of bone metabolism and disease activity in TNF-a inadequate responders with active rheumatoid arthritis. Akt Rheumatol 39: 370-374
-
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, et al. (2006) Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 203: 2673-2682.
-
Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, et al. (2000) T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 408: 600-605.
-
Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, et al. (2004) Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum 50: 650-659.
-
Lubberts E, Koenders MI, van den Berg WB (2005) The role of T-cell interleukin-17 in conducting destructive arthritis: lessons from animal models. Arthritis Res Ther 7: 29-37.
-
Liu XH, Kirschenbaum A, Yao S, Levine AC (2006) Interactive effect of interleukin-6 and prostaglandin E2 on osteoclastogenesis via the OPG/RANKL/RANK system. Ann N Y Acad Sci 1068: 225-233.
-
Wong PK, Quinn JM, Sims NA, van Nieuwenhuijze A, Campbell IK, et al. (2006) Interleukin-6 modulates production of T lymphocyte-derived cytokines in antigen-induced arthritis and drives inflammation-induced osteoclastogenesis. Arthritis Rheum 54: 158-168.
-
Baum R, Gravallese EM (2014) Impact of inflammation on the osteoblast in rheumatic diseases. Curr Osteoporos Rep 12: 9-16.
-
Monroe DG, McGee-Lawrence ME, Oursler MJ, Westendorf JJ (2012) Update on Wnt signaling in bone cell biology and bone disease. Gene 492: 1-18.
-
Yao W, Cheng Z, Shahnazari M, Dai W, Johnson ML, et al. (2010) Overexpression of secreted frizzled-related protein 1 inhibits bone formation and attenuates parathyroid hormone bone anabolic effects. J Bone Miner Res 25: 190-199.
-
Bodine PV, Zhao W, Kharode YP, Bex FJ, Lambert AJ, et al. (2004) The Wnt antagonist secreted frizzled-related protein-1 is a negative regulator of trabecular bone formation in adult mice. Mol Endocrinol 18: 1222-1237.
-
Morvan F, Boulukos K, Cl�ment-Lacroix P, Roman Roman S, Suc-Royer I, et al. (2006) Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res 21: 934-945.
-
Li J, Sarosi I, Cattley RC, Pretorius J, Asuncion F, et al. (2006) Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone 39: 754-766.
-
Uderhardt S, Diarra D, Katzenbeisser J, David JP, Zwerina J, et al. (2010) Blockade of Dickkopf (DKK)-1 induces fusion of sacroiliac joints. Ann Rheum Dis 69: 592-597.
-
Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, et al. (2007) Dickkopf-1 is a master regulator of joint remodeling. Nat Med 13: 156-163.
-
Wang SY, Liu YY, Ye H, Guo JP, Li R, et al. (2011) Circulating Dickkopf-1 is correlated with bone erosion and inflammation in rheumatoid arthritis. J Rheumatol 38: 821-827.
-
Heiland GR, Appel H, Poddubnyy D, Zwerina J, Hueber A, et al. (2012) High level of functional dickkopf-1 predicts protection from syndesmophyte formation in patients with ankylosing spondylitis. Ann Rheum Dis 71: 572-574.
-
Chen G, Deng C, Li YP (2012) TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci 8: 272-288.
-
Lories RJ, Derese I, Luyten FP (2005) Modulation of bone morphogenetic protein signaling inhibits the onset and progression of ankylosing enthesitis. J Clin Invest 115: 1571-1579.
-
Walsh NC, Gravallese EM (2010) Bone remodeling in rheumatic disease: a question of balance. Immunol Rev 233: 301-312.
-
Harre U, Georgess D, Bang H, Bozec A, Axmann R, et al. (2012) Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest 122: 1791-1802.
-
Neumann E, Frommer KW, Vasile M, M�ller-Ladner U (2011) Adipocytokines as driving forces in rheumatoid arthritis and related inflammatory diseases? Arthritis Rheum 63: 1159-1169.
-
Yamaguchi N, Kukita T, Li YJ, Kamio N, Fukumoto S, et al. (2008) Adiponectin inhibits induction of TNF-alpha/RANKL-stimulated NFATc1 via the AMPK signaling. FEBS Lett 582: 451-456.
-
Luo XH, Guo LJ, Xie H, Yuan LQ, Wu XP, et al. (2006) Adiponectin stimulates RANKL and inhibits OPG expression in human osteoblasts through the MAPK signaling pathway. J Bone Miner Res 21: 1648-1656.
-
M�ller-Ladner U, Neumann E (2009) Rheumatoid arthritis: the multifaceted role of adiponectin in inflammatory joint disease. Nat Rev Rheumatol 5: 659-660.
-
Thommesen L, Stunes AK, Monjo M, Gr�svik K, Tamburstuen MV, et al. (2006) Expression and regulation of resistin in osteoblasts and osteoclasts indicate a role in bone metabolism. J Cell Biochem 99: 824-834.
-
Meier FM, Frommer KW, Peters MA, Brentano F, Lef�vre S, et al. (2012) Visfatin/pre-B-cell colony-enhancing factor (PBEF), a proinflammatory and cell motility-changing factor in rheumatoid arthritis. J Biol Chem 287: 28378-28385.
-
Xie H, Tang SY, Luo XH, Huang J, Cui RR, et al. (2007) Insulin-like effects of visfatin on human osteoblasts. Calcif Tissue Int 80: 201-210.
-
Moschen AR, Geiger S, Gerner R, Tilg H (2010) Pre-B cell colony enhancing factor/NAMPT/visfatin and its role in inflammation-related bone disease. Mutat Res 690: 95-101.
-
Allali F, Breban M, Porcher R, Maillefert JF, Dougados M, et al. (2003) Increase in bone mineral density of patients with spondyloarthropathy treated with anti-tumour necrosis factor alpha. Ann Rheum Dis 62: 347-349.
-
Catrina AI, af Klint E, Ernestam S, Catrina SB, Makrygiannakis D, et al. (2006) Anti-tumor necrosis factor therapy increases synovial osteoprotegerin expression in rheumatoid arthritis. Arthritis Rheum 54: 76-81.
-
Dischereit G, Tarner IH, M�ller-Ladner U, Lange U (2013) Infliximab improves bone metabolism and bone mineral density in rheumatoid arthritis and ankylosing spondylitis: a prospective 2-year study. Clin Rheumatol 32: 377-381.
-
Lange U, Teichmann J, M�ller-Ladner U, Strunk J (2005) Increase in bone mineral density of patients with rheumatoid arthritis treated with anti-TNF-alpha antibody: a prospective open-label pilot study. Rheumatology (Oxford) 44: 1546-1548.
-
Vis M, Havaardsholm EA, Haugeberg G, Uhlig T, Voskuyl AE, et al. (2006) Evaluation of bone mineral density, bone metabolism, osteoprotegerin and receptor activator of the NFkappaB ligand serum levels during treatment with infliximab in patients with rheumatoid arthritis. Ann Rheum Dis 65: 1495-1499.
-
Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, et al. (2010) LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum 62: 929-939.
-
Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, et al. (2004) Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum 50: 1400-1411.
-
Lipsky PE, van der Heijde DM, St Clair EW, Furst DE, Breedveld FC, et al. (2000) Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med 343: 1594-1602.
-
Kensuke K, Amano K, Yamada S, Hatta K (2011) Tocilizumab monotherapy improves bone mineral density as well as TNF blockers plus methotrexate with methotrexate-resistant active rheumatoid arthritis. An Open-lable randomized clinical trial. T-bone Trial. Arthritis Rheum 63: 147-148.
-
Fleischmann RM, Halland AM, Brzosko M, Burgos-Vargas R, Mela C, et al. (2008) Tocilizumab Inhibits Structural Joint Damage in Rheumatoid Arthritis Patients with an Inadequate Response to Methotrexate: LITHE study 2-year results. J Rheumatol 40: 113-126
-
Smolen JS, Han C, van der Heijde DM, Emery P, Bathon JM, et al. (2009) Radiographic changes in rheumatoid arthritis patients attaining different disease activity states with methotrexate monotherapy and infliximab plus methotrexate: the impacts of remission and tumour necrosis factor blockade. Ann Rheum Dis 68: 823-827.
-
Smolen JS, Avila JC, Aletaha D (2012) Tocilizumab inhibit progression of joint damage in rheumatoid arthritis irrespective of its anti-inflammatory effects: disassociation of the link between inflammation and destruction. Ann Rheum Dis 71: 687-693.
-
Kim HR, Lee SH, Kim HY (2006) Elevated serum levels of soluble receptor activator of nuclear factors-kappaB ligand (sRANKL) and reduced bone mineral density in patients with ankylosing spondylitis (AS). Rheumatology (Oxford) 45: 1197-1200.
-
Dischereit G, Neumann N, M�ller-Ladner U, Lange U (2014) The impact of serial low-dose radon hyperthermia exposure on pain, disease activity and pivotal cytokines of bone metabolism in ankylosing spondylitis � a prospective study. Akt Rheumatol 39: 304-309.
-
Moder A, Hufnagel C, Lind-Albrecht G (2012) Effect of combined Low-Dose Radon- and HyperthermieTreatment (LDRnHT) of patients with ankylosing spondylitis on serum levels of cytokines and bone metabolism markers: a pilot study. Int J Low Radiation 7: 423-435.
-
Cohen SB, Dore RK, Lane NE, Ory PA, Peterfy CG, et al. (2008) Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum 58: 1299-1309.
-
Dore RK, Cohen SB, Lane NE, Palmer W, Shergy W, et al. (2010) Effects of denosumab on bone mineral density and bone turnover in patients with rheumatoid arthritis receiving concurrent glucocorticoids or bisphosphonates. Ann Rheum Dis 69: 872-875.
-
Genovese MC, Bathon JM, Martin RW, Fleischmann RM, Tesser JR, et al. (2002) Etanercept versus methotrexate in patients with early rheumatoid arthritis: two-year radiographic and clinical outcomes. Arthritis Rheum 46: 1443-1450.
-
Genovese MC, Durez P, Richards HB, Supronik, Jerzy D, et al. (2011) One year efficacy and safety results of a phase II trial of secukinumab in patients with rheumatoid arthritis. Arthritis Rheum 63: 147-148.
