

International Journal of Cancer and Clinical Research
A Simple Two-Stage PCR Method for Quality Amplification of Degenerate DNA
Ebili Henry O1*, Ham-Karim Hersh Abdul2 and Ilyas Mohammad2
1Department of Morbid Anatomy & Histopathology, Olabisi Onabanjo University, Nigeria
2Division of Pathology, University of Nottingham, Queen's Medical Centre, UK
*Corresponding author:
Ebili Henry O, Department of Morbid Anatomy & Histopathology, Olabisi Onabanjo University, Ago-Iwoye, Nigeria, Tel: +2348069629739, E-mail : bakabakaxl@yahoo.com
Int J Cancer Clin Res, IJCCR-2-018, ((Volume 2, Issue 3), Original Article; ISSN: 2378-3419
Received: June 05, 2015 | Accepted: July 03, 2015 | Published: July 06, 2015
Citation: Ebili HO, Ham-Karim HA, Ilyas M, Ilyas M (2015) A Simple Two-Stage PCR Method for Quality Amplification of Degenerate DNA. Int J Cancer Clin Res 2:018. 10.23937/2378-3419/2/3/1018
Copyright: © 2015 Ebili HO, et al . This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Formalin-fixed, paraffin-embedded (FFPE) tissue banks are invaluable to cancer genetics and biomarker discovery. However, FFPE tissue-derived DNA is degenerate. As such, any PCR technique which will give adequate amplification of such DNA to enable downstream applications is important to the characterization of genetic biomarkers and to personalized medicine. The aim of this article is to describe a two-stage PCR method which efficiently amplifies degenerate FFPE tissue-derived DNA. Amplicon-enriched templates were produced by a first-stage multiplex PCR which used five or more primer pairs with similar annealing temperature. Subsequently, the enriched products were used as templates for a singleplex PCR which utilized the same PCR cycling parameters and primer pairs as in the multiplex PCR. We compared the amplification of FFPE-derived and cell line DNA by this new protocol and the conventional PCR using the percentage of samples that achieved amplification at a cycle threshold (ct) of 30 or less, the mean of ct values obtained at the end of PCR, as well as our devised amplification efficiency test. Furthermore we compared the amplification of FFPE DNA using this present protocol and the Quick Multiplex Consensus-PCR (QMC-PCR) protocol on the fast programme of the ABI 7500 Fast Real Time PCR instrument. The present two-stage protocol has better sensitivity and amplification efficiency than the conventional PCR in amplifying degenerate DNA templates; and it also amplifies DNA better than the QMC-PCR protocol in fast cycling Real Time PCR programmes.
Keywords
Two-stage PCR, Quality amplification, Degenerate DNA
Abbreviations
FFPE: Formalin-Fixed Paraffin-Embedded, QMC-PCR: Quick Multiplex Consensus-PCR, HRMA: High Resolution Melt Analysis, NTC: No Template Control, ct: cycle threshold
Introduction
Across the world histopathology departments maintain a rich tissue bank in the form of formalin-fixed, paraffin-embedded (FFPE) tissues for medical diagnosis and research; and for a long time to come FFPE tissue will continue to be the most available and reliable method of preserving tissue architecture and biomolecules, at least in developing countries [1-3]. FFPE tissue banks are therefore an invaluable source of data for biomedical research [1-3]. Currently, the discovery and characterization of novel genetic biomarkers are proving relevant to personalized treatment of human diseases [4,5].
However, DNA derived from FFPE tissue is degenerate and difficult to amplify by PCR, the commonest method of testing for genetic biomarkers [4,5]. This is because formalin fixation damages DNA by cross-linking DNA strands [1-3]. This damage is made worse when fixation conditions have been suboptimal and when samples have been stored for long periods of time [1]. In many histopathology archives some FFPE samples are over 50 years old [1,2]. This provides significant challenges for the process of discovery of genetic biomarkers as the DNA from these FFPE tissues are suboptimal for PCR analysis [1-3]. Therefore, any technique which aids the PCR amplification of FFPE DNA in order to utilize it in downstream applications is most desirable.
Previously, our research group described the Quick Multiplex Consensus PCR (QMC-PCR) for obtaining high quality amplification from degenerate DNA template for downstream applications such as High resolution Melt Analysis (HRMA) and pyrosequencing [6,7]. Here we describe a modification of the QMC-PCR protocol. This method is a two-stage PCR protocol consisting of an initial multiplex PCR which produces amplicon-enriched products. These products are subsequently used as templates in a singleplex PCR. The same primer pairs and cycling parameters as in the multiplex PCR are used in the singleplex PCRs. We found that this protocol amplifies significantly more FFPE tissue-derived DNA samples earlier and more efficiently than the conventional PCR. We also found that in a fast PCR programme, the two-stage PCR amplifies degenerate DNA better than the QMC-PCR method.
Materials and Methods
A total of 89 samples were included in this study. This comprised 78 DNA samples from FFPE tissues (76 gastric cancer tissues and 2 normal tissues) obtained from the archives of the Pathology Department of the University College Hospital, Ibadan, Nigeria. These gastric cancer tissues were 3 to 34 years old in the UCH Pathology archives, and consisted of gastrectomy specimens, gastric endoscopic biopsies, omental nodules of metastatic gastric cancer tissue and lymph node biopsies. The remaining samples included 1 normal FFPE tissue-derived DNA, 4 FFPE colorectal cancer tissue-derived DNA and DNA from 7 colorectal cancer cell lines all obtained from the stocks of Pathology Research Group of the University of Nottingham, United Kingdom. All the PCRs were run in the ABI 7500 Fast Real Time PCR machine.
DNA extraction
Four to eight shavings of the UCH, Ibadan, FFPE samples were subjected to DNA extraction using the Promega ReliaPrep™ FFPE gDNA Miniprep System kit according to the manufacturer's instruction.
The two-stage PCR protocol
The first stage: The following genetic regions were amplified for each samples using multiplex PCR: KRAS exons 2, 3 and 4; BRAF exons 11 and 15; PIK3CA exons 9 and 20; using the primers shown in table 1. All the primers were shown to have an annealing temperature of about 55℃ using gradient PCR. The cycling parameters include 3 minutes of initial denaturation at 95℃, 40-45 cycles consisting of 10 seconds of denaturation at 95℃, 45 seconds of annealing at 55℃ and 30 seconds of extension at 72℃; followed by 5 minutes of final extension at 72℃. PCR was carried out in a 10μl volume made up of 5μl of Diamond Hotshot master mix, 1μl of LC Green Plus dye, 300nM of primers and 40ng DNA template.
![]()
Table 1: Sequences of primer pairs used for amplifications of the genes in this study
View Table 1
At the end of the PCR 1:100 dilutions of the products were made by adding 4μl of PCR products to 396μl of distilled water. The diluted products were used as DNA template for the second stage reactions.
The second stage: Using 300nM of KRAS exon 3 primer pairs, 2μl of diluted products as DNA template and all other parameters from the first stage a singleplex PCR was used to amplify exon 3 of KRAS.
Conventional PCR protocol
Conventional PCR consisting of 3 cycling steps of denaturation, annealing and extension were carried out on 40ng (or more, see Results below) of FFPE DNA using the same cycling parameters above; and the results were compared with that of the two-stage PCR protocols described above.
Criteria for adequate amplification
The criteria used to adjudge quality and adequate amplification were those that have been previously described: cycle threshold (ct) value of 30 or less and amplification curve with exponential segment and amplitude (Applied Biosystems High Resolution Melting Getting Started Guide www.appliedbiosystems/hrm) [8].
Efficiency testing for conventional versus two-stage PCR
To compare the efficiency of the conventional PCR and that of our 2-stage PCR, four serial 1:10 dilutions (up to 1:1000) of original FFPE DNA and amplicon-enriched DNA template, both of which are from the same sample stock, were prepared by adding 5μl of template to 45μl of water. Two microliters of each of the dilutions were subsequently used in PCR which was run in duplicates utilizing the parameters described above. The original FFPE DNA amount for the conventional PCR was 40ng. The Excel spreadsheet (2010) was used to plot the graphs of log10 of serial dilutions versus ct values and obtain the amplifications slopes. The values of the slopes were subsequently used to derive the amplification efficiencies of both PCR protocols.
The Quick Multiplex Consensus (QMC) protocol versus the two-stage PCR protocol
Amplification of exon 10 of the POL E gene was used to compare the QMC-PCR protocol as previously described and the two-stage PCR protocol on the ABI Fast Real Time PCR using the Fast PCR module. The primer sequences used for this study are shown in table 1.
Sequencing
The PCR products of the two-stage PCR were sequenced using the Sanger method. The chromatograms were viewed and interpreted with the Finch TV software.
Statistical analysis
The student t independent test (SPSS version 19) was used to determine the mean ct values and differences of mean ct values between the conventional and two-stage PCR protocols.
Results
Amplification Quality: conventional PCR versus two-stage PCR protocols
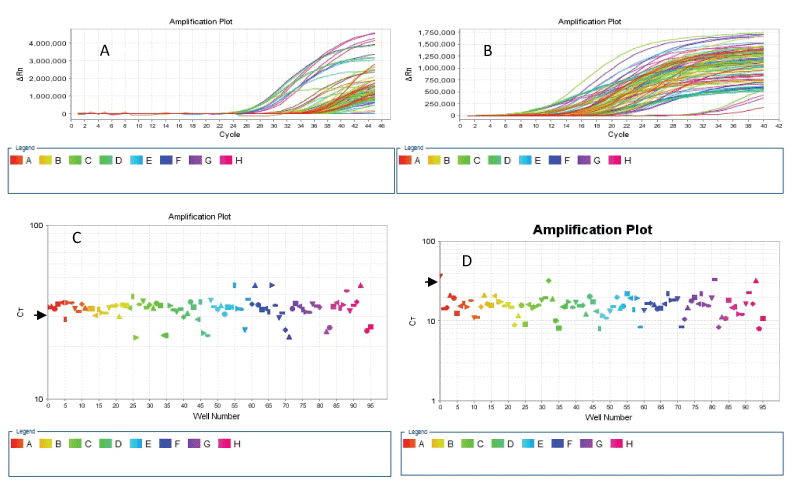
We compared the amplification of exon 3 of KRAS in about 94 samples (comprising singles of 76 gastric cancer derived DNA and duplicates of 2 normal DNA and 7 colorectal cancer cell lines-derived DNA) amplified by the conventional PCR and the two-stage PCR protocols. Using the following criteria for adequate amplification: cycle threshold (ct) value of 30 or less and an amplification curve with exponential phase and amplitude, we found that while 92/94 (97.9%) of the samples achieved amplification at ct values of less than 30 when using the two-stage PCR protocol, only about 16/94 (17%) of the samples (all cell line DNA and DNA from normal tissue) attained the criteria of ct of 30 or less when the conventional PCR protocol was used (Figure 1).
.
Figure 1: Linear graph of ΔRN vs Cycle (A & B) and log graph of CT vs Well (C & D) show amplification of KRAS exon 3 in 96 sample wells. The 96 samples wells comprise DNA of 76 FFPE gastric cancer tissue samples, duplicate samples of 2 normal FFPE tissues, duplicates DNA samples from 7 colorectal cancer cell lines and 2 no-template controls (NTCs). The ΔRN vs Cycle plots show that most samples amplified by the conventional PCR technique (A) failed to achieve amplification at the ct value of 30; while all except 4 samples (including the 2 NTC samples) show adequate amplification at a ct value of 30 or less when the two-stage PCR protocol was used (B). The CT vs Wells plots show that only 17 of the 96 wells were amplified at a ct value of 30 (short arrow) or less with the conventional PCR (C), in contrast to the two-stage PCR results (D) which show that 92 wells were amplified adequately.
View Figure 1
Amplification 'sensitivity': conventional PCR versus two-stage PCR protocols
We hypothesized that using higher amount of FFPE DNA templates and higher primer concentrations in a conventional PCR may give similar quality amplification as the two-stage PCR protocol. We tested this hypothesis by amplifying 80ng of original templates of 18 odd samples with 500nM of primers for the conventional PCR, while maintaining the same template amount and primer concentration as used previously for the amplicon-enriched group. Using the student t independent test we found that the difference in mean ct value of samples between the conventional and the two-stage protocol was 23.24 ± 0.62 (95% confidence interval of 21.99 - 24.50; p<0.001; conventional PCR=29.60 ± 2.29, two-stage PCR=6.35 ± 1.22). This result shows that the two-stage PCR is more 'sensitive' in amplifying degenerate templates such as FFPE DNA.
Amplification efficiency: conventional PCR versus two-stage PCR protocols
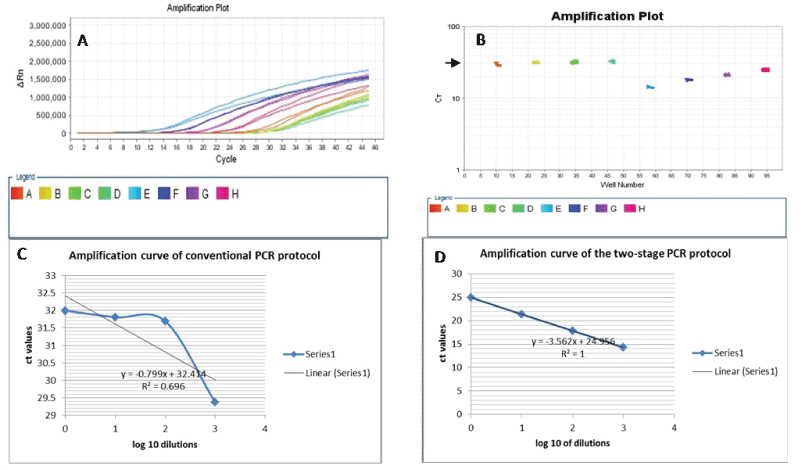
Duplicate samples of 4 serial 1:10 dilutions (up to 1:1000) of original FFPE DNA template and amplicon-enriched DNA template, both of which are from the same sample stock were subjected to conventional 3-step PCR (denaturation, annealing and extension) of KRAS exon 3. As shown in table 2 there was good replication of all the dilutions in the 2 groups of samples. But while all the serial dilutions that were amplified by the two-stage PCR protocol achieved adequate amplifications, only the 1:1 replicates of the original templates (conventional PCR) achieved a ct value of about 30 or less (Table 2). The other dilutions did not achieve the adequate amplification criteria. Furthermore, we tested the amplification efficiencies of conventional and two-stage PCR for our FFPE DNA samples by deriving a standard curve for both protocols using the averages of the ct values and log10 of dilution in Excel 2010. We then used the values of the slopes in the equation, E=0(-1/s) -1, where E is amplification efficiency and s is the slope of the derived curve (Figure 2). The amplification efficiency of the conventional PCR protocol was 1678%, while the two-stage PCR protocol achieved 90.9% efficiency at amplifying our set of samples.
.
Figure 2: A. Amplification of KRAS exon3 from serial dilutions of original DNA and amplicon-enriched DNA templates, both from the same patient sample. B. Each of the dilution levels of both template types was perfectly replicated (conventional PCR: wells 11& 12, 23 & 24, 35 & 36, 47 & 48; two-stage PCR: wells 59&60, 71 & 72, 83 & 84, 95&96). However, only the 1:1 replicates of the original DNA template was amplified at the CT value of 30 (well 11 & 12 at short arrow in B). On the other hand, all the dilution series of the amplicon-enriched templates adequately amplified, achieving amplification at less than 30 ct value. C and D. Standard curves for the amplification of FFPE DNA samples using the conventional versus the two-stage PCR. The two-stage PCR shows a better efficiency at amplifying degenerate (FFPE) DNA.
View Figure 2
![]()
Table 2: Ct values of template dilutions amplified by the conventional and the two-stage pcr
View Table 2
The QMC-PCR protocol versus the two-stage PCR protocol
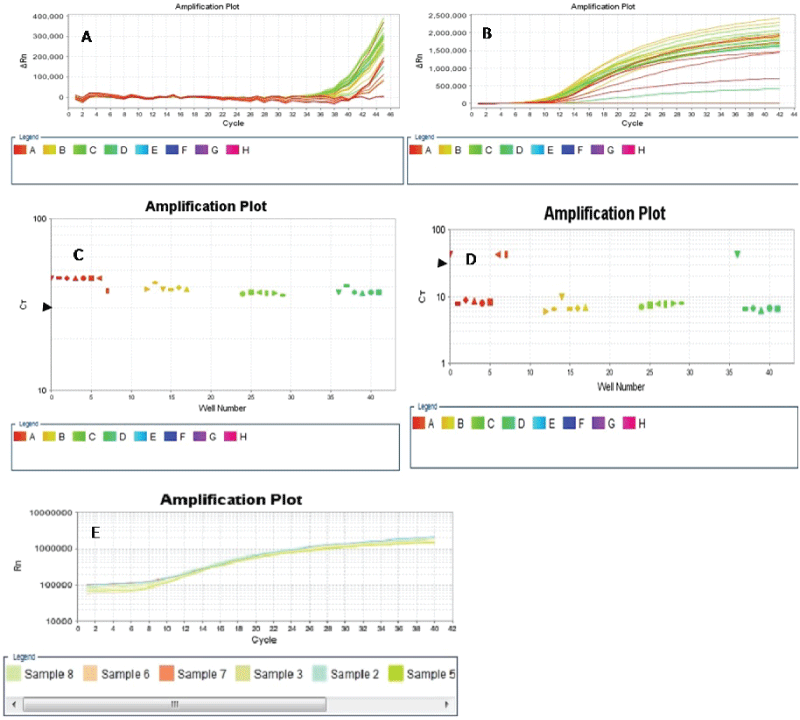
We compared the two-stage PCR protocol with the QMC protocol on the ABI 7500 Fast Real Time PCR machine by using the Fast PCR programme to amplify exon 10 of POL E-enriched FFPE colorectal cancer-derived templates prepared from the multiplex stage of the QMC-PCR protocols. Our results showed that while the two-stage PCR protocol adequately amplified the templates, the QMC protocol did not (Figure 3). This suggested that the two-stage PCR protocol may be superior to the QMC protocol when using a high ramping speed programme such as the ABI 7500 Fast programme. We also utilized a two-step PCR process consisting of 10 seconds of denaturation and 45 seconds of annealing on the ABI fast programme to amplify exon 10 of POL E-enriched colorectal cancer-derived DNA. The amplification of POL E using this two-step PCR achieved adequate amplification (Figure 3E). We concluded that the poor amplification obtained with the QMC-PCR protocol on the high ramp speed programme was due to the significantly reduced reaction times of the QMC-PCR protocol; and that when using a high speed ramp instrument such as the ABI 7500 Fast machine our two-stage PCR protocol may be superior to the QMC-PCR protocol previously described by our group.
.
Figure 3: Four samples were amplified in six replicates at exon 10 of the POL E gene using high speed ramping on the ABI 7500 Real Time PCR machine. While the QMC-PCR protocol produced poor amplification across all the samples and their replicates (A & C), amplification with the two-stage PCR protocol on the fast programme was excellent (B & D), with the exception of wells A1 and D1 (D) which kinked during the PCR run and did not achieve the ct of 30 (arrowheads) or the normal amplification curve (B). Wells A7 and A8 were the NTCs. E. Amplification of POL E-enriched templates using a two-step cycling with denaturation time of 10 seconds and 45seconds annealing time. Amplification was better with our two-stage method than with the QMC-PCR.
View Figure 3
Specificity of the two-stage PCR method
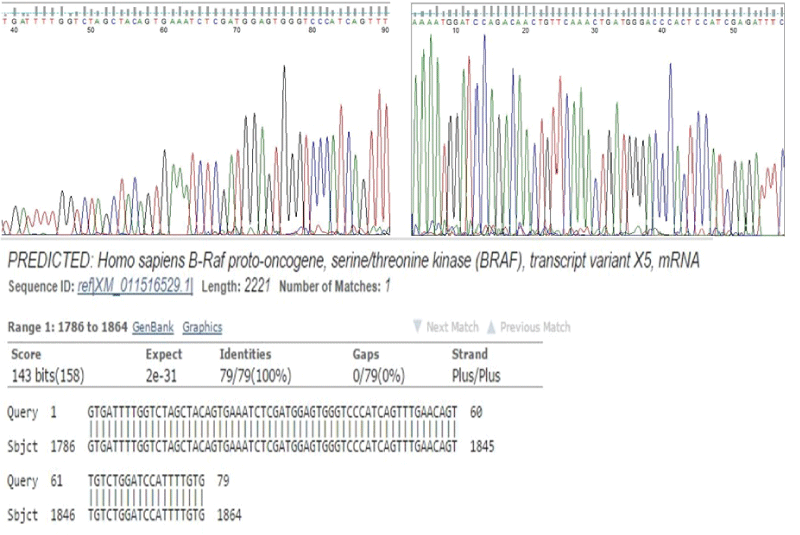
In order to determine the specificity of the two-stage PCR method, i.e. whether it introduces errors during the amplification process or not, BRAF exon 15 sequence was amplified in FFPE tissue-derived DNA using the two-stage PCR method. The PCR products were sequenced using the Sanger method. Visual inspection of the obtained forward and reverse sequences showed that there were no changes to the target sequence. Also, when the obtained sequences were queried on BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi), they showed a perfect match to the target BRAF exon 15 sequence in the databases (Figure 4).
.
Figure 4: Sanger sequencing of the two-stage PCR products. A and B. Chromatogram of Sanger forward (A) and reverse (B) sequences of the two-stage PCR BRAF exon 15 amplification product. Visual inspection of the chromatogram shows that there was no artifactitious insertion or deletion of nucleotides in the sequences. C. BLAST analysis of the Sanger sequences show perfect matches with database sequences.
View Figure 4
Discussion
We have shown here that the two-stage PCR protocol is clearly superior to conventional PCR in amplifying degenerate DNA such as FFPE DNA. Across a large bulk of samples, the two-stage protocol adequately amplified almost all the samples, while more than half of the samples gave poor quality amplification with conventional PCR.
Hilscher et al had previously used the mean ct value and the differences between mean ct values as a measure of the sensitivity of the PCR assay to detect viral nucleic acid [9]. In this study we adopted the mean ct value and difference in mean ct value between the two PCR protocols as a measure of the ability of the PCR protocol to amplify degenerate DNA template. We showed that the two-stage PCR protocol achieves early amplification of the genes of interest from FFPE DNA, in contrast to the conventional PCR which is characterized by late amplifications. The practical implication of our results is that much more than the conventional protocol, the two-stage PCR protocol is robust in amplifying degenerate DNA templates.
Furthermore, similar to the measure used by Hilscher et al. we have used the standard curve derived from serial dilutions of samples in determining the amplification efficiency [9]. And we showed that, like the 'sensitivity', the two-stage PCR has an acceptable efficiency status of 90.9% (range is 90-105% [Bio-Rad Real Time PCR Application Guide www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_5279.pdf]) at amplifying our set of FFPE DNA than the conventional protocol.
We have also shown that this two-stage PCR method produces specific PCR products. Sequencing of the two-stage PCR-derived products revealed no changes in the product sequence. This means that the two-stage PCR method reliably amplifies DNA sequences from degenerate template such as FFPE DNA and finds application in HRM analysis and screening of point mutations (manuscripts in preparation). Other PCR-based downstream application where this protocol may be applicable include mutation-specific PCR (allele-specific PCR), methylation-specific PCR and methylation-sensitive HRM [10-12]. However, our protocol may not be applicable to the comparative quantitative PCR as differences in the amplification efficiencies between primers for the gene(s) of interest and the house-keeping gene may give spurious gene dosage results over the two amplification stages [13].
This protocol may find use in fields such as Forensic Pathology and Archaeology where the only template available may be degenerate [14,15].
The QMC-PCR protocol has previously been shown to be superior to conventional PCR in amplifying degenerate FFPE DNA ; and has been applied to HRM and sequencing with excellent results [6,7] The differences between the QMC and this new protocol are (i) while the QMC uses nested primers in the second stage, this protocol uses the same primer pairs for both stages of the amplification; (ii) in the QMC-PCR method the second stage PCR is a two-step PCR consisting of 1 second of denaturation and 1-10 seconds of annealing, but in the two-stage protocol, the singleplex (second) stage amplification is either two- or three-step PCR cycling with reaction times in the conventional PCR range [6].
While both the QMC-PCR and the present two-stage methods worked well when using standard ramp speed (data not shown), we showed here that with the fast PCR ramp speed the QMC-PCR method produces poor amplification of the final template. The reason for this poor amplification with the fast cycling PCR on fast ramping may be due to the significantly reduced time the reaction spends on each cycle (which is a combination of the denaturation, annealing and total ramping times). In the study by Hilscher et al. it was also noted that fast PCR conditions were associated with loss of sensitivity (or loss of ability to amplify the DNA of interest) and it was suggested that with fast PCR cycling conditions fewer amplicons are produced per cycle [9].
Although the concept of amplicon-enrichment for subsequent PCR is not new, and has been described for QMC-PCR, whole genome amplification and other two-stage amplification methods, the technique we describe here is simple requiring no complex primer or probe design or cycling protocol. It uses the same primer pairs and cycling parameters for both stages of the protocol [6,7,17-20].
Finally, further manipulation of the two-stage protocol can reduce experiment time and/or the amount of primers in the singleplex stage of the protocol. For example, using lower dilutions such as 1:20 or 1:50, rather than 1:100 produces more concentrated amplicons of interest in the dilutions. This can be exploited to reduce the number of PCR cycles in the second stage (Figure 5) and /or the primer concentrations (not done). This description of the two-stage PCR method assumes that one is amplifying multiple gene regions. However, the first stage of the two-stage PCR can be a singleplex reaction if one is interested in amplifying only one gene region from the FFPE DNA.
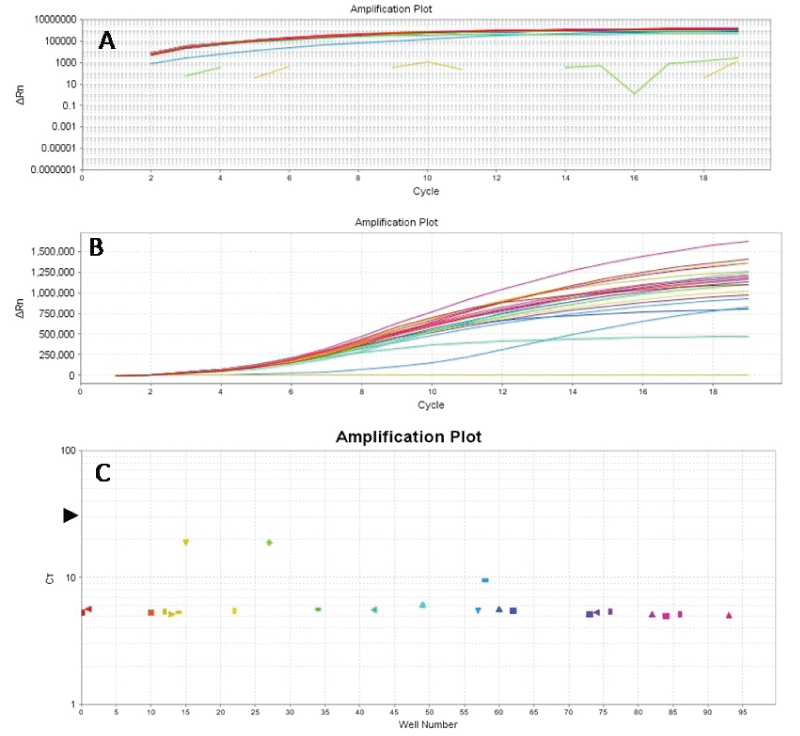
.
Figure 5: Amplification plots (Log graph of ΔRN vs Cycle (A), linear graph of ΔRN vs Cycle (B) and CT vs Wells (C)) of BRAF exon 15. BRAF exon 15 was amplified from 40ng of original DNA templates of FFPE gastric cancer tissues using the same primer concentrations and cycling parameters referred to in the text. Following amplification, 1:20 dilutions of the products were prepared, and 2μl of these were used in the second stage PCR utilizing the same primer concentrations and cycling parameters, except that the number of cycles was reduced to 19 from 40-45 cycles. The results show that adequate amplifications were achieved for the samples at ct values of 4-9.5 (not all samples are shown here) and about 20 for the NTCs (4C); and a reduction of PCR run time of about 1hour 10 minutes due to reduction in cycle number.
View Figure 5
In conclusion we have described a two-stage PCR protocol, a modification of the QMC-PCR which is superior to conventional PCR in amplifying degenerate FFPE DNA. In addition we showed that this two-stage PCR protocol may have some advantage over the fast cycling QMC-PCR protocol on a high speed ramping instrument.
Acknowledgement
We thank the Department of Pathology, University College Hospital, Ibadan, for providing some of the samples for the study, and Prof EEU Akang of the Department of Pathology, UCH, Ibadan for proofreading the manuscript. We wish to acknowledge the Pathological Society of Great Britain and Ireland which provided the grant for this study, as well as the Union for International Control of Cancer for the Travel and Maintenance award provided HOE for the study. Finally, we acknowledge Drs W. Fadhil and S. Ibrahem who designed most of the primers used for this study.
References
-
Michalik SS (2008) Overcoming poor quality DNA. Drug Discovery Dev 11: 24-30.
-
Srinivasan M, Sedmak D, Jewell S (2002) Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am J Pathol 161: 1961-1971.
-
Lin J, Kennedy SH, Svarovsky T, Rogers J, Kemnitz JW, et al. (2009) High-quality genomic DNA extraction from formalin-fixed and paraffin-embedded samples deparaffinized using mineral oil. Anal Biochem 395: 265-267.
-
Clark-Langone KM, Wu JY, Sangli C, Chen A, Snable JL, et al. (2007) Biomarker discovery for colon cancer using a 761 gene RT-PCR assay. BMC Genomics 8: 279.
-
5. Li L, Fridley BL, Kalari K, Niu N, Jenkins G, et al. (2014) Discovery of genetic biomarkers contributing to variation in drug response of cytidine analogues using human lymphoblastoid cell lines. BMC Genomics 15: 93.
-
Fadhil W, Ibrahem S, Seth R, Ilyas M (2010) Quick-multiplex-consensus (QMC)-PCR followed by high-resolution melting: a simple and robust method for mutation detection in formalin-fixed paraffin-embedded tissue. J Clin Pathol 63: 134-140.
-
Ibrahem S, Seth R, O'Sullivan B, Fadhil W, Taniere P, et al. (2010) Comparative analysis of pyrosequencing and QMC-PCR in conjunction with high resolution melting for KRAS/BRAF mutation detection. Int J Exp Pathol 91: 500-505.
-
White H, Potts G (2006) Mutation scanning by high resolution melt analysis. Evaluation of rotor-gene 6000 (Corbett Life Science), HR-1 and 384-well Lightscanner (Idaho Technology) National Genetics Reference Laboratory (Wessex) (NGRLW_HRM_1.0): 1-45
-
Hilscher C, Vahrson W, Dittmer DP (2005) Faster quantitative real-time PCR protocols may lose sensitivity and show increased variability. Nucleic Acids Res 33: e182.
-
Lang AH, Drexel H, Geller-Rhomberg S, Stark N, Winder T, et al. (2011) Optimized allele-specific real-time PCR assays for the detection of common mutations in KRAS and BRAF. J Mol Diagn 13: 23-28.
-
Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A 93: 9821-9826.
-
Wojdacz TK, Dobrovic A (2007) Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res 35: e41.
-
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101-1108.
-
Holland MM, Cave CA, Holland CA, Bille TW (2003) Development of a quality, high throughput DNA analysis procedure for skeletal samples to assist with the identification of victims from the World Trade Center attacks. Croat Med J 44: 264-272.
-
Gugerli F, Parducci L, Petit RJ (2005) Ancient plant DNA: review and prospects. New Phytol 166: 409-418.
-
Zheng Y-M, Wang N, Li L, Jin F (2011) Whole genome amplification in preimplantation genetic diagnosis. Journal of Zhejiang University-SCIENCE B 2: 1-11.
-
Kusser WC, Levin DB, Glickman BW (1993) Sensitive two-stage PCR of p53 genomic DNA exons 5-9. PCR Methods Appl 2: 250-252.
-
Beauchamp NJ, Daly ME, Cooper PC, Preston FE, Peake IR (1994) Rapid two-stage PCR for detecting factor V G1691A mutation. Lancet 344: 694-695.
-
Durmaza R, Aydinb A, Durmaza B, Aydinb NE, Akbasakc BS, et al. (1997) Sensitivity of two-stage PCR amplification for detection of Mycobacterium tuberculosis in paraffin-embedded tissues. Journal of Microbiological Methods 29: 69-75.
-
Kamal IH, Al Gashgari B, Moselhy SS, Kumosani TA, Abulnaja KO (2013) Two-stage PCR assay for detection of human brucellosis in endemic areas. BMC Infect Dis 13: 145.
