

International Journal of Cancer and Clinical Research
Acetylation of α-tubulin by a Histone Deacetylase Inhibitor, Resminostat, Leads Synergistic Antitumor Effect with Docetaxel in Non-Small Cell Lung Cancer Models
Hiroaki Konishi1*, Akimitsu Takagi1, Hiroyuki Takahashi2, Satoru Ishii2, Yu Inutake2 and Takeshi Matsuzaki1
1Yakult Central Institute, Yakult Honsha Co., Ltd., Japan
2Pharmaceutical Research and Development Department, Yakult Honsha Co., Ltd., Japan
*Corresponding author:
Hiroaki Konishi, Yakult Central Institute, Yakult Honsha Co., Ltd., 5-11 Izumi, Kunitachi-shi, Tokyo 1860-8650, Japan, Tel: +81-42-577-8960, Fax: +81-42-577-3020, E-mail: hiroaki-konishi@yakult.co.jp
Int J Cancer Clin Res, IJCCR-4-077, (Volume 4, Issue 1), Original Article; ISSN: 2378-3419
Received: January 10, 2017 | Accepted: January 27, 2017 | Published: January 31, 2017
Citation: Konishi H, Takagi A, Takahashi H, Ishii S, Inutake Y, et al. (2017) Acetylation of α-tubulin by a Histone Deacetylase Inhibitor, Resminostat, Leads Synergistic Antitumor Effect with Docetaxel in Non-Small Cell Lung Cancer Models. Int J Cancer Clin Res 4:077. 10.23937/2378-3419/1410077
Copyright: © 2017 Konishi H, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Background: There is a growing body of clinical evidence to demonstrate that inhibition of histone deacetylase is effective in the treatment of various types of cancer. We examined whether acetylation of a non-histone protein α-tubulin was induced by resminostat and this acetylation exerts combination effects with docetaxel since α-tubulin was a target of docetaxel.
Methods: The cytotoxicity of resminostat was evaluated in 11 human non-small cell lung cancer cell lines and the inhibitory activity of resminostat against histone deacetylase 6 was also determined. The stability of α-tubulin (the amount of polymerized and acetylated α-tubulin) was analyzed in NCI-H460 cells treated with resminostat and/or docetaxel. Caspase-3/7 activity was analyzed in NCI-H460, A549, NCI-H1975, and MOR/CPR cell lines treated with resminostat alone or in combination with docetaxel. The antitumor efficacies of resminostat and docetaxel were evaluated inxenograft mouse models.
Results: Resminostat exhibited anti-proliferative activity in all 11 cell lines tested (IC50, 0.36-8.7 μmol/L) and also inhibited histone deacetylase 6 activities. The amounts of polymerized and acetylated α-tubulin and caspase-3/7 activity were increased by resminostat alone or the combination with docetaxel. This combination synergistically increased antitumor efficacy in all the tumor-bearing mice tested.
Conclusion: These results demonstrate that the combination of resminostat and docetaxel offers a promising regimen in the treatment of non-small cell lung cancer.
Keywords
Histone deacetylase inhibitor, Non-small cell lung cancer, Combination chemotherapy, Docetaxel, Acetylation of α-tubulin
Introduction
Lung cancer is the leading cause of cancer-related death worldwide [1]. A number of pharmacological agents are frequently used in the treatment of non-small cell lung cancer (NSCLC), including cisplatin (CDDP), docetaxel (DTX), and erlotinib, an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) [2]. Among them, DTX inhibits the depolymerization of microtubules. This stabilizing effect is then followed by an increase in the polymerization of α-tubulin [3]. Monotherapy with DTX, however, has proven insufficient to produce a satisfactory response; hence clinical study is focusing on its effect in combination with other agents.
One such agent is a histone deacetylase (HDAC) inhibitor. Inhibition of HDAC activity is followed by the accumulation of acetylated histones and acetylated non-histone proteins. HDAC regulates chromatin remodeling and is crucial in the reactivation of epigenetically silenced tumor suppressor genes [4,5]. Inhibitors of HDAC have been reported to mediate transcriptional activity by modulating the acetylation status of nucleosomal histones proximal to gene promoters [6]. In addition to chromatin remodeling, HDAC activity (mainly HDAC6) has beenlinked to the acetylation of non-histone proteins such as tumor suppressor p53 and α-tubulin [7-9]. Excessive acetylation of α-tubulin leads to the stabilization of microtubules, resulting in cytotoxicity [10].
HDAC inhibitors and DTX stabilize microtubules in different ways [3,10]. This suggests that combination therapy with HDAC inhibitors and DTX might achieve a therapeutic effect on NSCLC from the mode of action not only of efficacy, but also of safety. We examined the efficacy of combination treatment with resminostat [11-13], an HDAC inhibitor, and DTX in four chemotherapy-resistant NSCLC cell lines: A549, NCI-H460, NCI-H1975, and MOR/CPR. The first two harbor an amino acid substitution on KRAS (G12S and Q61H, respectively), which affects activation ofthe MAPK and PI3K-AKT pathways [14]; NCI-H1975 cells harbor a T790M substitution on EGFR that renders cancer cells resistant to EGFR-TKIs [15]; and MOR/CPR cells are CDDP-resistant due to reduced accumulation of platinum complexes [16]. These cell lines represent typical chemotherapy-resistant mechanisms in NSCLC cells. One problem with combination therapies is that although they offer an increase in efficacy, they also mean a compromise in safety due to a tendency toward an increase in adverse effects. One toxic side effect of DTX is myelosuppression [17], whereas with resminostat, gastrointestinal disturbances are more common [12]. This suggests that combination therapy with resminostat and DTX might offer a potent therapy against NSCLC, while avoiding synergistic adverse effects. In addition, DTX-based therapy enhances the stabilization of microtubules through a different biochemical mechanism than resminostat, which acetylates α-tubulin, suggesting a potential improvement in therapeutic efficacy.
The purpose of this study was to investigate the efficacy of combination therapy with resminostat and DTX against chemotherapy-resistant NSCLC cells. Our hypothesis was that this combination might provide one of novel regimens for the clinical treatment of NSCLC.
Methods
Reagents and cell cultures
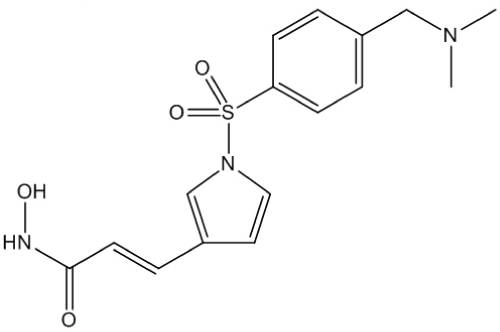
Resminostat (Figure 1) was obtained from Yakult Honsha Co., Ltd. (Tokyo, Japan). DTX (TAXOTERE®) was obtained from Sanofi K.K. (Tokyo, Japan). Suberoylanilide hydroxamic acid (SAHA) was obtained from Alexis Corporation (Lausen, Switzerland) and tubacin from Selleck Chemicals (Houston, TX, USA). Human NSCLC cell linesA549, NCI-H460, and NCI-H1975 were obtained from ATCC (Manassas, VA, USA). Another human NSCLC cell line, MOR/CPR, was obtained from the European Collection of Cell Cultures (Salisbury, UK). All the cell lines were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO, USA) and antibiotics. The absence of mycoplasma contamination was confirmed periodically with the MycoAlertTM Mycoplasma Detection Kit (Lonza, Basel, Switzerland).
Inhibition of HDAC activity in NCI-H460 cells
The NCI-H460 cells were treated with resminostat or SAHA for 60 min to determine the level of inhibitory activity of each of them against HDAC I/II. The HDAC I/II activity in these cells were subsequently determined using the HDAC-Glo™ I/II Assay and Screening System (Promega, Madison, WI, USA). The inhibitory activity of SAHA or resminostat against HDAC6 was also determined with the HDAC6 Fluorimetric Drug Discovery Kit (Enzo Life Sciences Inc., Farmingdale, NY, USA).
Anti-proliferative activity
The cells (A549, NCI-H460, NCI-H1975, and MOR/CPR) were seeded into a 96-well culture plate (2500 cells/well; 50 μL/well) and resminostat and/or DTX added the next day. The number of viable cells was determined after 48 h incubation using the Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan).
Cell cycle in NCI-H460 cells
The NCI-H460 cells were exposed to resminostat (2-50 μmol/L) for 24 h. The cell cycle in these cells (5000 cells) was analyzed using theCell Cycle Phase Determination Kit (Cayman Chemical Company, Ann Arbor, MI, USA) and a Guava easy CyteTM flow cytometer (Merck Millipore, Darmstadt, Germany).
Immunoblotting of cell cycle- and apoptosis-related proteinsin NCI-H460 cells
In the cell cycle-related protein analysis, the NCI-H460 cells were treated with resminostat (20 μmol/L) for1-24 h; in the apoptosis-related protein analysis, they were treated with resminostat (10 μmol/L) and/or DTX (10, 20 nmol/L) for 24 h. After treatment, the cells were harvested and lysed in RIPA buffer (10 mmol/L Tris-HCl, pH7.4; 0.1% nonidet-P-40; 0.1% sodium deoxycholate; 0.1% SDS; 150 mmol/L NaCl; 1 mmol/L EDTA; 10 μg/mL aprotinin; and a phosphatase inhibitor cocktail) (Nacalai Tesque, Inc., Kyoto, Japan). Equal amounts of protein were subjected to SDS-PAGE on an 8-15% gel. The separated proteins were transferred to a nitrocellulose membrane. The membrane was incubated with the following primary antibodies: anti-Acetyl-Histone H3 (CST-9677; Cell Signaling Technology, Inc. CST, Beverly, MA, USA), anti-Histone H3 (CST-9715; CST), anti-p21 (sc-397; Santa Cruz Biotechnology, Dallas, TX, USA), anti-Cyclin D1 (CST-2926; CST), anti-phospho-retinoblastoma (pRB) (CST-9307; CST), anti-RB (CST-9302;CST), anti-Cyclin B1 (sc-752; Santa Cruz Biotechnology), anti-α-tubulin (T9026; Sigma-Aldrich), anti-acetylated-α-tubulin (T6793; Sigma-Aldrich), anti-X-linked inhibitor of apoptosis protein (XIAP)(CST-2045; CST), anti-B-cell lymphoma 2(BCL-2) (sc-492; Santa Cruz Biotechnology), anti-Bcl-2-like protein 11 (BIM)(CST-2933; CST), and anti-β-actin (A5316; Sigma-Aldrich). All the antibodies were used at a dilution of 1:3000. Immune complexes were detected with horseradish peroxidase-conjugated antibodies (Sigma-Aldrich) and chemiluminescence reagents (GE Healthcare, Buckinghamshire, UK). All other chemicals were of the highest purity available.
Immunoblotting of polymerized α-tubulinin NCI-H460 cells
The NCI-H460 cells were treated with resminostat (10 μmol/L), tubacin (10 μmol/L), and/or DTX (2 nmol/L) for 24 h. The cells were then suspended in lysis buffer (20 mmol/L Tris-HCl, pH7.4; 0.5% nonidet-P-40; 1 mmol/L MgCl2.6H2O; 2 mmol/L EGTA; 10 μg/mL aprotinin; and a phosphatase inhibitor cocktail) for detection of polymerized α-tubulin. Cells lysates were clarified by centrifugation at 20,000 g for 10 min at 4°C. The precipitate was lysed in RIPA buffer. For detection of other proteins (listed in Figure 2), the cells were lysed in RIPA buffer.
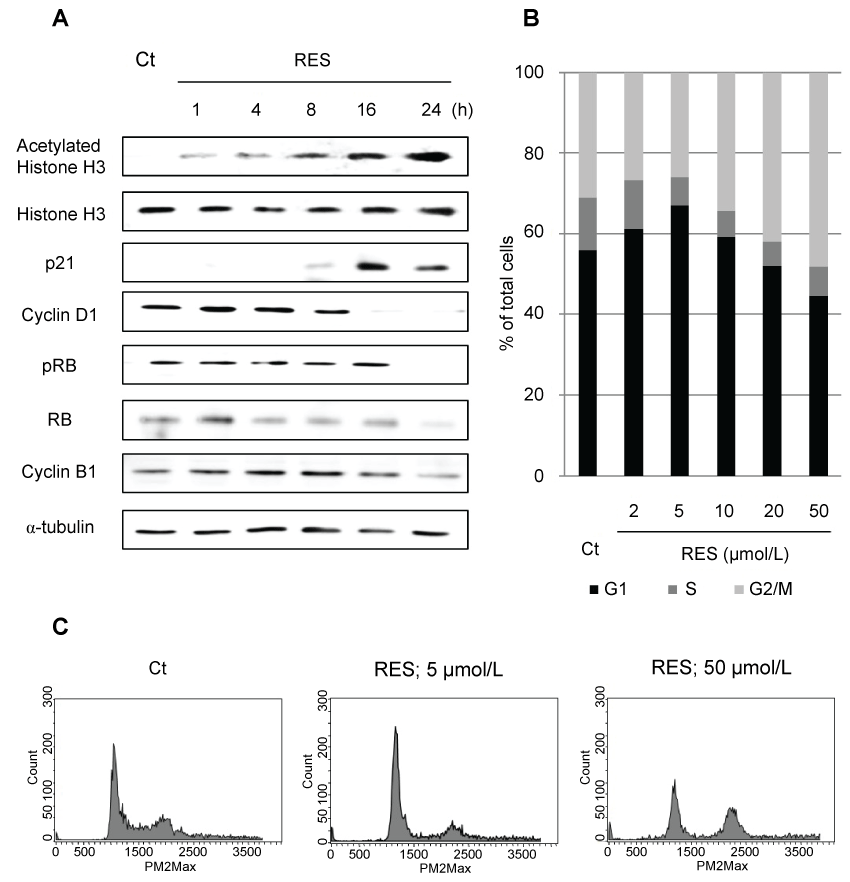
.
Figure 2: Levels of polymerized and acetylated α-tubulin after treatment with RES, tubacin, or docetaxel (DTX). NCI-H460 cells were treated with RES (10 μmol/L), tubacin (10 μmol/L), and/or DTX (2 nmol/L) as indicated for 24 h. Amounts of polymerized and acetylated α-tubulin were determined by immunoblotting.
View Figure 2
Caspase-3/7 activity
The cells (A549, NCI-H460, NCI-H1975, and MOR/CPR) were seeded into a 96-well, white-walled plate (2500 cells/well; 50 μL/well) and resminostat and/or DTX added the next day. Caspase-3/7 activity was determined after 24 h incubation using Caspase-GloTM 3/7 Assays (Promega).
Subcutaneous xenograft models
Five-week-old male BALB/c-nu/nu mice were obtained from Japan SLC, Inc. (Hamamatsu, Japan). Cultured tumor cells (2 × 106 cells/mouse) were injected subcutaneously into the flank of each mouse. Each tumor lesion was measured in two dimensions by caliper twice a week and volume calculated using the following formula: tumor volume (mm3) = 1/2 × (major axis) × (minor axis)2. When tumor volume reached 100-200 mm3, the mice were randomized and treated with resminostat (60 or 90 mg/kg/dose, oral administration (p.o.), 5 times/week, formulated with 0.5% methylcellulose in water) and/or DTX (5 or 10 mg/kg/dose, intravenous administration (i.v.), once every 3 weeks, formulated under the manufacturer's instructions).
Statistical analysis
Group differences were analyzed by using a one-way analysis of variance (Dunnett).
Results
Resminostat suppresses proliferation of NSCLC cells
Resminostat and SAHA showed similar inhibitory activity against HDAC I/II (cell-based assay) and HDAC6 (enzyme assay) (Table 1). The cytotoxic activity of resminostat was determined in NSCLC cells, including those with a KRAS mutation and chemotherapy-resistance (Table 2). The average IC50 value for the cytotoxicity of resminostat against the KRAS wild type cell line was 4.9 μmol/L (range 2.0-8.7 μmol/L), while that against the KRAS mutation cell line was 2.9 μmol/L (range 0.36-4.5 μmol/L) (Table 2).
![]()
Table 1: HDAC inhibitory activity of resminostat (RES).
View Table 1
![]()
Table 2: IC50 values for resminostat anti-proliferative activity against NSCLC cells.
View Table 2
Additionally, no cross-resistance to resminostat was observed in EGFR-TKI-or CDDP-resistant cells. Resminostat-resistant NSCLC cells were not detected in 11 cell lines. The influence of resminostat on the cell cycle and related proteins wasalso determined to investigate the relationship between re-expressed proteins and cell cycle arrest. Resminostat induced expression of p21 accompanied by acetylation of histone H3. Subsequent degradation of cyclin D1 and dephosphorylation of RB ensued (Figure 3A). The cell cycle analysis revealed that resminostat induced G1 phase arrest at lower (2-5 μmol/L) and G2/M phase arrest at higher concentrations (20-50 μmol/L) (Figure 3B and Figure 3C). The status of the cell cycle and related proteins changed depending on the concentration used and duration of treatment.
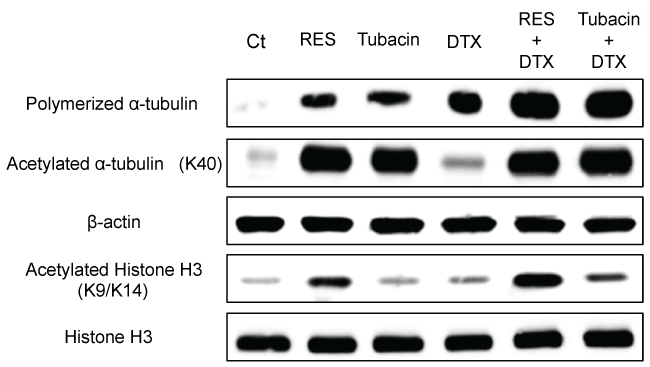
.
Figure 3: Change in cell cycle-related protein levels and cell cycle profiles after treatment with resminostat (RES). (A) Immunoblotting analysis of cell cycle-related proteins. NCI-H460 cells were treated with RES (20 μmol/L) for1-24 h. After treatment, cell cycle-related proteins were examined by immunoblotting; (B) Cell cycle profile; (C) Typical cell cycle histograms. NCI-H460 cells were treated with RES (2-50 μmol/L) for 24 h. After treatment, cell cycle statuses of 5000 cells were determined.
View Figure 3
Combination of resminostat and DTX synergistically promoted stabilization of microtubules and apoptosis
Excessive stabilization of microtubules leads to the suppression of cell division. The stabilization of α-tubulin in resminostat and/or DTX-treated cells was determined with respect to the polymerization and acetylation status of α-tubulin. Resminostat induced acetylation of α-tubulin and promoted its polymerization to the same extent as tubacin, a selective HDAC6 inhibitor. DTX induced polymerization of α-tubulin but not its acetylation. Combination treatment promoted polymerization to a greater extent than resminostat or DTX alone (Figure 3). Next, the effects of stabilization on apoptosis were examined. Expression of anti/pro-apoptotic proteins XIAP, BCL-2, and BIM, and caspase-3/7 activity were determined as indicators of induction of apoptosis. Resminostat decreased expression of XIAP and BCL-2, and induced expression of BIM (Figure 4A). Treatment with resminostat or DTX alone induced caspase-3/7 activity in all the tested NSCLC cells; in A549 cells, however, this effect was observed at higher concentrations (data not shown). In addition, combination treatment induced higher caspase-3/7 activity in NCI-H460, A549, and NCI-H1975 cells, than single-agent therapy, and this difference was significant (Figure 4B).
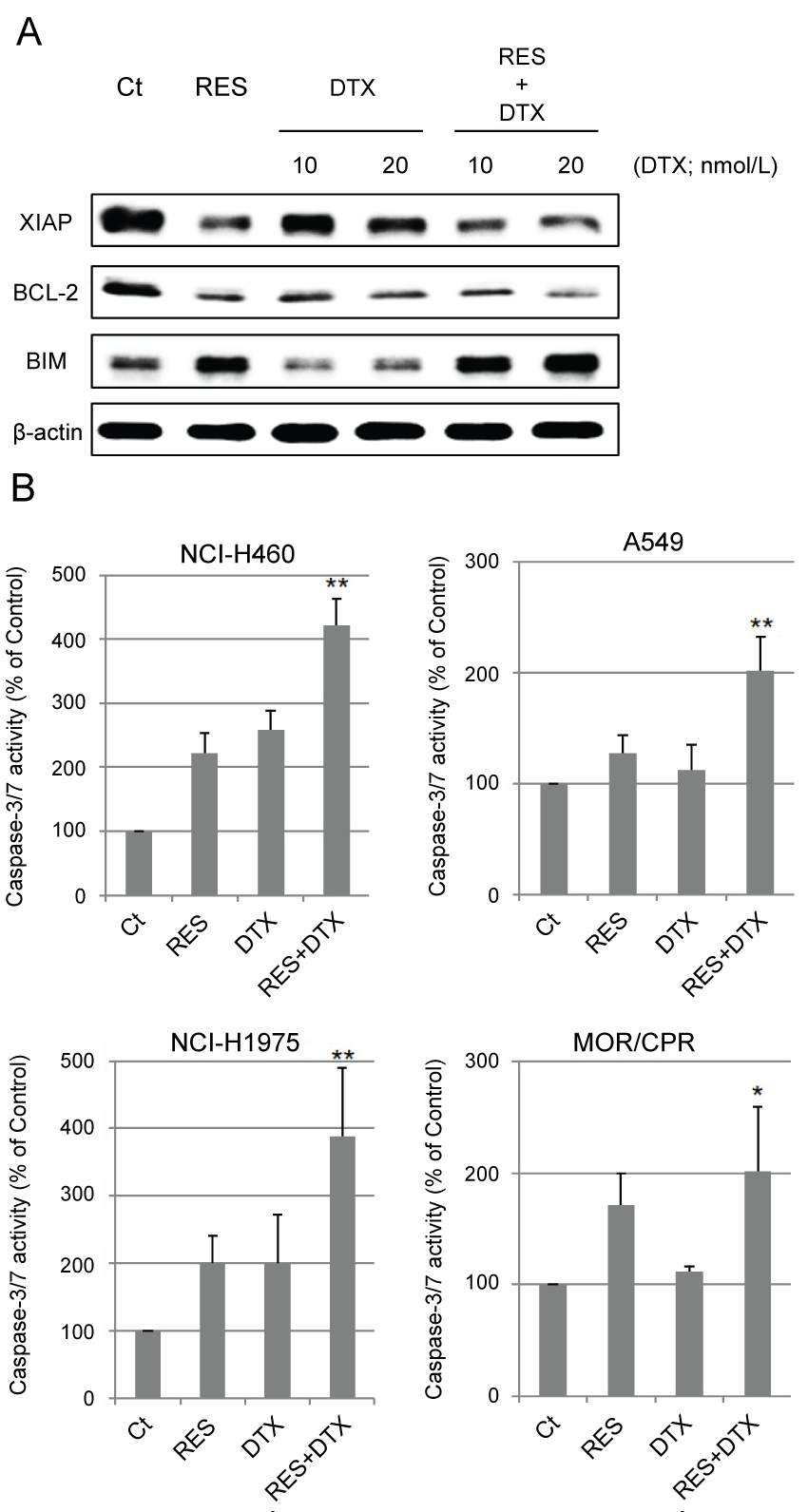
.
Figure 4: Change in apoptosis-related proteins and caspase-3/7 activity. (A) Immunoblotting analysis. NCI-H460 cells were treated with RES (10 μmol/L) and/or DTX (10, 20 nmol/L) for 24 h. After treatment, amounts of XIAP, BCL-2, and BIM were determined by immunoblotting; (B) Activity of Caspase-3/7. Four NSCLC cell lines were treated with RES 10 μmol/L (NCI-H460 cells), 2.5 μmol/L (A549 and NCI-H1975 cells), 0.5 μmol/L (MOR/CPR cells), and/or DTX 10 nmol/L (NCI-H460, A549, and NCI-H1975 cells) or 1 nmol/L (MOR/CPR cells) for 24 h. Apoptotic cells were detected as those exhibiting caspase-3/7 activity (calculated from 3 independent experiments).
*p < 0.05 vs. Control, **p < 0.01 vs. Control and single agents.
View Figure 4
Combination of resminostat and DTX inhibits tumor growth in chemotherapy-resistant NSCLC cells in nude mice
The in vivo efficacy of resminostat in combination with DTX was subsequently determined. The antitumor efficacy was determined in two different schedules, intermittent administration schedule and daily administration schedule. In the former schedule, the mice were treated with resminostat on days 1-5, 8-12, 22-25, 29-33 (Figure 5A). In the latter schedule, the mice were treated with resminostat on days 1-5, 8-12, 15-19 (Figure 5B). There was no difference on antitumor efficacy. Monotherapy with resminostat or DTX yielded a moderate decrease in tumor growth, whereas combination therapy resulted in an appreciable improvement in antitumor efficacy in all the NSCLC tumors tested (Figure 5B- 5E) ( p < 0.05 vs. Control). None of the mice treated with these agents showed any severe macroscopic adverse effects.
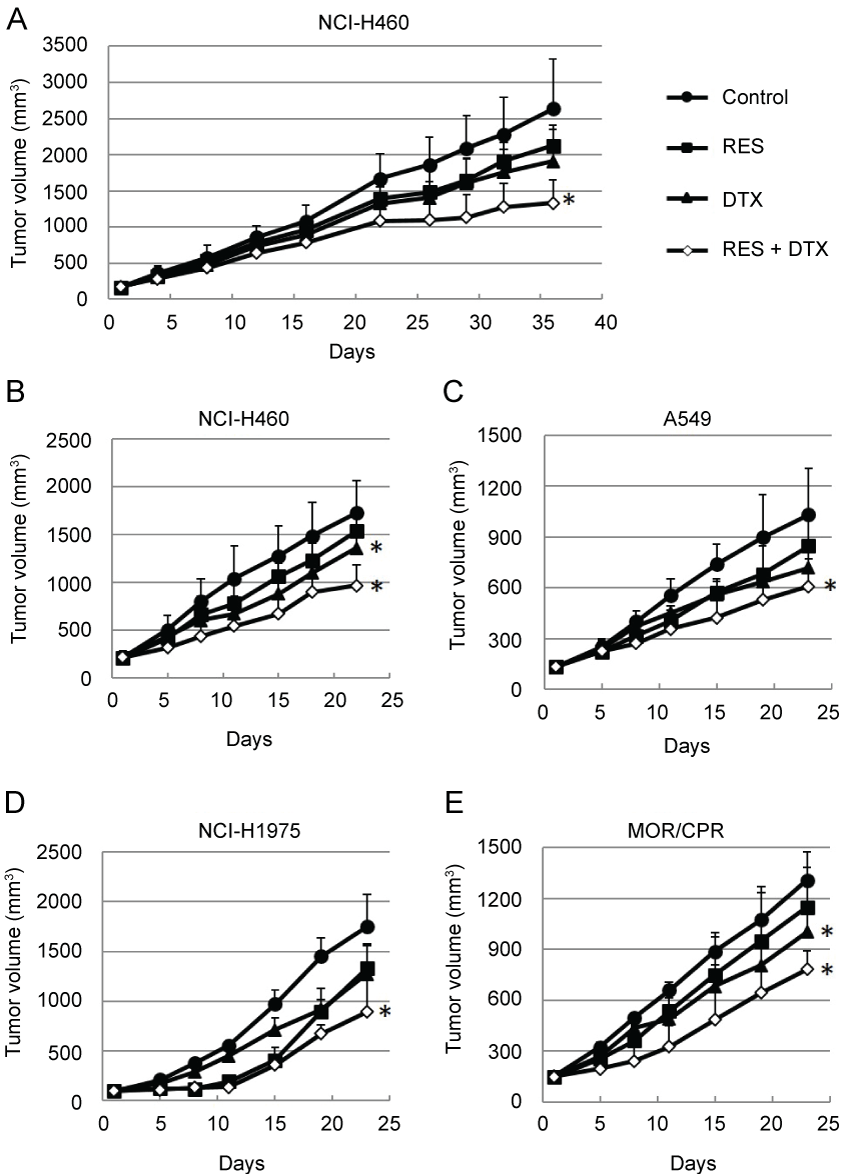
.
Figure 5: Antitumor efficacies of RES and/or DTX in mouse xenograft models of non-small cell lung cancers. (A) Nude mice bearing NCI-H460 tumors were treated with RES (90 mg/kg) on days 1-5, 8-12, 22-26, and 29-33 and/or DTX (5 mg/kg) on days 1 and 29; (B-E) Nude mice bearing (B) NCI-H460; (C) A549; (D) NCI-H1975; or (E) MOR/CPR tumors were treated with RES 90 mg/kg/dose (NCI-H460, NCI-H1975, and MOR/CPR tumors) on days 1-5, 8-12, and 15-19 and/or DTX 10 mg/kg/dose (NCI-H460 and A549 tumors) or 5 mg/kg/dose (NCI-H1975 and MOR/CPR tumors) on day 1. Tumor volume was measured using calipers on days indicated. Mean ± SD tumor volumes are shown for groups of 5 mice. *p < 0.05 vs. Control.
View Figure 5
Discussion
The present results revealed that combination therapy with resminostat and DTX was effective againstchemotherapy-resistant NSCLC cells. This combination therapy exerted a synergistic effect, resulting in an increase in antitumor efficacy against several NSCLC cells both in vitro and in vivo, especially with respect to the stabilization of microtubules. Resminostat exhibitsits cytotoxicity on chemotherapy-resistant NSCLC cells through various mechanisms. In this study, no cross-resistance to resminostat was observed in EGFR-TKI-or CDDP-resistant or KRAS mutation cells. The difference in the cytotoxic effect of resminostat and DTX is likely due to the mode of action of each reagent. In general, tubulin-targeting agents exert strong cytotoxicity, whereas HDAC inhibitors exert moderate cytotoxicity [18,19]. HDAC inhibitors have several modes of action, including tubulin acetylation, HSP90 inhibition, and pro-apoptosis protein re-expression [20-23]. An agent with several modes of action is less likely to induced resistance due to specific mechanisms. In fact, in the present study, no cross-resistance to resminostat was detected in other chemotherapy-resistant cell lines. This suggests that the combination of resminostat and DTX could be effective in the treatment of NSCLC.
Combination therapy with resminostat and DTX appears to be a suitable optionwith respect to its mode of action, i.e., the stabilization of microtubules. Increased levels of acetylated α-tubulin with resminostat might be due to inhibition of HDAC6 (Table 1). Acetylated α-tubulin contributes to the stabilization of microtubules, whereas DTX inhibits α-tubulin depolymerization. Thus, used in combination, these two agents exert a synergistic effect in increasing polymerization of α-tubulin, resulting in its stabilization. In addition, these results correlated with those of the apoptosis experiments, which indicated synergistic activation of caspase-3/7.
Resminostat suppressed the proliferation of tumor cells in the G1 phase, and DTX killed other cells in the G2/M phase through stabilization of microtubules. Resminostat induced p21 expression, which was followed by a decrease in cyclin D1 expression and phosphorylation of RB. This arrest of the G1 phase may have been due to this decrease in phospho-RB and cyclin D1 [24-27]. The stabilization of α-tubulin had a significant influence on the cell cycle, resulting in arrest at the G2/M phase [28]. In the present study, acetylation of α-tubulin led to stabilization of microtubules, resulting in G2/M phase arrest in cells treated with high concentrations of resminostat. In an earlier study, resminostat induced G1 phase arrest in 3 out of 4 myeloma cell lines associated with upregulation of p21, and G2/M phase arrest in 2 out of 4 cell lines at different concentrations. The cause of this G2/M phase arrest remains to be identified [11]. However, resminostat exhibits multiple modes of action, including induction of p21 expression and acetylation of α-tubulin. Thus, both G1 and G2/M phase arrest induced by resminostat appears to be due to its exposure conditions.
The combination of resminostat and DTX exerted a synergistic antitumor effect against chemotherapy-resistant NSCLC cells in both the in vitro and in vivo studies. Resminostat alone increased expression of pro-apoptotic while decreasing expression of anti-apoptotic proteins. Induction of BIM expression might be due to enhancement of FOXO3A and E2F1 promoter activity [29,30]. It has been reported that low BIM expression can lead to resistance to TKI treatment [31]. In the present study, induction of BIM expression may have led to synergistic cytotoxicity against chemotherapy-resistant cell lines. Reduced expression of BCL-2 may be caused by decreased binding of its transcription factor to the BCL-2 promoter due to reduced interaction between the transcription factor and HDAC2 [32]. Change in apoptosis-related proteins influences the cytotoxicity of many antitumor agents. This indicates that resminostat might offer potent synergistic efficacy when used in combination with other antitumor agents. In the present in vivo study, resminostat combination therapy synergistically enhanced antitumor efficacy independent of the dosing schedule and cell line used. The cell lines used here represent several chemotherapy-resistant properties in clinical practice [14-16]. The in vivo studies validated the effectiveness of the in vitro combination studies with respect to cytotoxicity and tubulin stabilization assays. No severe toxicity was observed in the mice with any of the agents used in the present study. Therefore, resminostat combination therapy is regarded as well tolerated. These results demonstrated that resminostat combination therapy offers a feasible option for NSCLC patients.
Conclusions
Our study indicates that a rational strategy in combination with resminostat and DTX is thought to a promising regimen in the treatment of chemothrapy-resistant NSCLC. Based on the present results, we are proceeding with a phase I/II study in NSCLC patients to further investigate the safety and efficacy of thisregimen.
Acknowledgement
The authors thank Mrs. Rika Morita for her skilled technical assistance, Dr. Rolf Krauss (4SC AG) for meaningful discussion and constructive criticism to draft of this paper, and Drs. Yoshiyuki Shishido and Momomi Tsugane (Yakult Central Institute) for comments on this paper.
Conflict of Interest
The authors have no conflict of interest.
Ethical Statement
All the in vivo experimental protocols were approved by the Animal Care Committee of the Yakult Central Institute prior to the study.
References
-
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, et al. (2015) Global cancer statistics, 2012. CA Cancer J Clin 65: 87-108.
-
Ho C, Tong KM, Ramsden K, Ionescu DN, Laskin J (2015) Histologic classification of non-small-cell lung cancer over time: reducing the rates of not-otherwise-specified. Curr Oncol 22: e164-170.
-
Ringel I, Horwitz SB (1991) Studies with RP 56976 (taxotere): a semisynthetic analogue of taxol. J Natl Cancer Inst 83: 288-291.
-
Xu WS, Parmigiani RB, Marks PA (2007) Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 26: 5541-5552.
-
Hirose T, Sowa Y, Takahashi S, Saito S, Yasuda C, et al. (2003) p53-independent induction of Gadd45 by histone deacetylase inhibitor: coordinate regulation by transcription factors Oct-1 and NF-Y. Oncogene 22: 7762-7773.
-
Gui CY, Ngo L, Xu WS, Richon VM, Marks PA (2004) Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci U S A 101: 1241-1246.
-
Luo J, Su F, Chen D, Shiloh A, Gu W (2000) Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408: 377-381.
-
Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL (2003) Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A 100: 4389-4394.
-
Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, et al. (2002) HDAC6 is a microtubule-associated deacetylase. Nature 417: 455-458.
-
Murphy ME, Cassimeris L (2006) A novel cancer therapy approach targeting microtubule function. Cancer Biol Ther 5: 1721-1723.
-
Mandl-Weber S, Meinel FG, Jankowsky R, Oduncu F, Schmidmaier R, et al. (2010) The novel inhibitor of histone deacetylase resminostat (RAS2410) inhibits proliferation and induces apoptosis in multiple myeloma (MM) cells. Br J Haematol 149: 518-528.
-
Brunetto AT, Ang JE, Lal R, Olmos D, Molife LR, et al. (2013) First-in-human, pharmacokinetic and pharmacodynamic phase I study of Resminostat, an oral histone deacetylase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 19: 5494-5504.
-
Kitazono S, Fujiwara Y, Nakamichi S, Mizugaki H, Nokihara H, et al. (2015) A phase I study of resminostat in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol 75: 1155-1161.
-
Martin P, Leighl NB, Tsao MS, Shepherd FA (2013) KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J Thorac Oncol 8: 530-542.
-
Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, et al. (2005) EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 352: 786-792.
-
Twentyman PR, Wright KA, Mistry P, Kelland LR, Murrer BA (1992) Sensitivity to novel platinum compounds of panels of human lung cancer cell lines with acquired and inherent resistance to cisplatin. Cancer Res 52: 5674-5680.
-
Fossella FV, DeVore R, Kerr RN, Crawford J, Natale RR, et al. (2000) Randomized phase III trial of docetaxel versus vinorelbine or ifosfamide in patients with advanced non-small-cell lung cancer previously treated with platinum-containing chemotherapy regimens. The TAX 320 Non-Small Cell Lung Cancer Study Group. J Clin Oncol 18: 2354-2362.
-
Chang J, Varghese DS, Gillam MC, Peyton M, Modi B, et al. (2012) Differential response of cancer cells to HDAC inhibitors trichostatin A and depsipeptide. Br J Cancer 106: 116-125.
-
Samadi N, Ghanbari P, Mohseni M, Tabasinezhad M, Sharifi S, et al. (2014) Combination therapy increases the efficacy of docetaxel, vinblastine and tamoxifen in cancer cells. J Cancer Res Ther 10: 715-721.
-
Fiskus W, Ren Y, Mohapatra A, Bali P, Mandawat A, et al. (2007) Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-alpha levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin Cancer Res 13: 4882-4890.
-
Nakagawa T, Takeuchi S, Yamada T, Ebi H, Sano T, et al. (2013) EGFR-TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC inhibition. Cancer Res 73: 2428-2434.
-
Kurundkar D, Srivastava RK, Chaudhary SC, Ballestas ME, Kopelovich L, et al. (2013) Vorinostat, an HDAC inhibitor attenuates epidermoid squamous cell carcinoma growth by dampening mTOR signaling pathway in a human xenograft murine model. Toxicol Appl Pharmacol 266: 233-244.
-
Lee SJ, Hwang SO, Noh EJ, Kim DU, Nam M, et al. (2014) Transactivation of bad by vorinostat-induced acetylated p53 enhances doxorubicin-induced cytotoxicity in cervical cancer cells. Exp Mol Med 46: e76.
-
DeCaprio JA, Ludlow JW, Lynch D, Furukawa Y, Griffin J, et al. (1989) The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell 58: 1085-1095.
-
Mihara K, Cao XR, Yen A, Chandler S, Driscoll B, et al. (1989) Cell cycle-dependent regulation of phosphorylation of the human retinoblastoma gene product. Science 246: 1300-1303.
-
Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, et al. (1994) D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol 14: 2066-2076.
-
Sherr CJ (2004) Principles of tumor suppression. Cell 116: 235-246.
-
Hernandez-Vargas H, Palacios J, Moreno-Bueno G (2007) Molecular profiling of docetaxel cytotoxicity in breast cancer cells: uncoupling of aberrant mitosis and apoptosis. Oncogene 26: 2902-2913.
-
Essafi A, Fernandez de Mattos S, Hassen YA, Soeiro I, Mufti GJ, et al. (2005) Direct transcriptional regulation of Bim by FoxO3a mediates STI571-induced apoptosis in Bcr-Abl-expressing cells. Oncogene 24: 2317-2329.
-
Biswas SC, Liu DX, Greene LA (2005) Bim is a direct target of a neuronal E2F-dependent apoptotic pathway. J Neurosci 25: 8349-8358.
-
Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, et al. (2012) A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med 18: 521-528.
-
Duan H, Heckman CA, Boxer LM (2005) Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol 25: 1608-1619.
