

Journal of Rheumatic Diseases and Treatment
Aspirin-Triggered Resolvin D1 Versus Dexamethasone in the Treatment of Sjögren's Syndrome-Like NOD/ShiLtJ Mice - A Pilot Study
Justin T Easley, Joel W Nelson, Rachel E Mellas, Salah Sommakia, Chunhua Wu, Bryan Trump, and Olga J Baker*
School of Dentistry, University of Utah, Salt Lake City, UT 84108-1201, USA
*Corresponding author: Olga J. Baker, DDS, PhD, School of Dentistry, University of Utah, 383 Colorow Dr. Room 289A, Salt Lake City, UT 84108-1201, Tel: 801-587-1773, Fax: 801-585-6712, E-mail: olga.baker@hsc.utah.edu
J Rheum Dis Treat, JRDT-1-027, (Volume 1, Issue 4), Research Article; ISSN: 2469-5726
Received: September 28, 2015 | Accepted: December 15, 2015 | Published: December 17, 2015
Citation: Easley JT, Nelson JW, Mellas RE, Sommakia S, Wu C, et al. (2015) Aspirin-Triggered Resolvin D1 Versus Dexamethasone in the Treatment of Sjögren's Syndrome-Like NOD/ShiLtJ Mice - A Pilot Study. J Rheum Dis Treat 1:027. 10.23937/2469-5726/1510027
Copyright: © 2015 Easley JT, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Resolvin D1 (RvD1) and its aspirin-triggered epimeric form (AT-RvD1) are endogenous lipid mediators (derived from docosahexaenoic acid, DHA) that control the duration and magnitude of inflammation in models of complex diseases. Our previous studies demonstrated that RvD1-mediated signaling pathways are expressed and active in salivary glands from rodents and humans. Furthermore, treatment of salivary cells with RvD1 blocked TNF- -mediated inflammatory signals and improved epithelial integrity. The purpose of this pilot study was to determine the feasibility of treatment with AT-RvD1 versus dexamethasone (DEX) on inflammation (i.e., lymphocytic infiltration, cytokine expression and apoptosis) observed in submandibular glands (SMG) from the NOD/ShiLtJ Sjögren's syndrome (SS) mouse model before experimenting with a larger population. NOD/ShiLtJ mice were treated intravenously with NaCl (0.9%, negative control), AT-RvD1 (0.01-0.1 mg/kg) or DEX (4.125-8.25 mg/kg) twice a week for 14 weeks beginning at 4 weeks of age. At 18 weeks of age, SMG were collected for pathological analysis and detection of SS-associated inflammatory genes. The AT-RvD1 treatment alone did not affect lymphocytic infiltration seen in NOD/ShiLtJ mice while DEX partially prevented lymphocytic infiltration. Interestingly, both AT-RvD1 and DEX caused downregulation of SS-associated inflammatory genes and reduction of apoptosis. Results from this pilot study suggest that a systemic treatment with AT-RvD1 and DEX alone attenuated inflammatory responses observed in the NOD/ShiLtJ mice; therefore, they may be considered as potential therapeutic tools in treating SS patients when used alone or in combination.
Keywords
Resolvins, Salivary Glands, AT-RvD1, RvD1, Sjögren's syndrome, Submandibular Gland, Cytokine
Introduction
Sjögren's Syndrome (SS) is an autoimmune disorder characterized by chronic inflammation of the salivary and lacrimal glands, resulting in secretory dysfunction [1]. Previous studies have found elevated levels of pro-inflammatory cytokines in salivary glands from SS patients [2] and SS mouse models [3]. Specifically, tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), interleukin-1β (IL-1β), interleukin-6 (IL-6), interleukin-18 (IL-18), interleukin-12 (IL-12), BAFF, interleukin-17 (IL-17) and interleukin-23 (IL-23) are significantly elevated [2-5]. Interestingly, the anti-inflammatory cytokines such as transforming growth factor beta 1 (TGFβ1) and interleukin-4 (IL-4) were expressed at low levels but interleukin-10 (IL-10) was overexpressed (for review, please see reference [5]). The etiology of SS remains unknown and treatment options are limited to either secretory agonists such as pilocarpine or saliva substitutes [6].
Previous studies demonstrated that human and animal cells convert ω-3 polyunsaturated fatty acids (PUFAs) into resolvins (Rv), which are anti-inflammatory and pro-resolving agents that control the duration and magnitude of inflammation and promote tissue repair [7]. Rv subtypes include the E-series (RvE1-2, derived from eicosapentaenoic acid [EPA]), the D-series (RvD1 and RvD2, derived from docosahexaenoic acid [DHA]), and aspirin-triggered forms (AT-RvD1) [8]. Rvs naturally inhibit key immuno-inflammatory processes in response to infection, injury or environmental challenges [9]. Interestingly, they are able to preserve tissue integrity and promote tissue repair and regeneration following an environmental insult [10-12]. Our previous studies indicate that ALX/FPR2, the receptor for RvD1, is expressed and active in the rat parotid cell line, Par-C10 [13]. Specifically, activation of ALX/FPR2 with AT-RvD1 blocked inflammatory signals caused by TNF-α and enhanced salivary epithelial integrity [13]. ALX/FPR2 is also expressed in mouse submandibular gland (SMG), and its activation with AT-RvD1 increases a diverse set of intracellular signaling pathways (e.g., intracellular calcium [Ca2+]i, Erk1/2, and Akt) to block TNF-α-mediated caspase-3 activation [14]. We also demonstrated that the machinery for RvD1 biosynthesis and its receptor are expressed and functional in human minor salivary glands (hMSG) with and without SS [15]. The expression levels of ALX/FPR2 seem to be unaltered in hMSG with and without SS [15], indicating that RvD1 could be used as a therapeutic option to treat salivary gland inflammation in SS patients.
One of the current treatments for SS includes the use of oral corticosteroids (see reviews [6,16]) and parotid irrigation with corticosteroids [17]. Additionally, dexamethasone (DEX) has been previously shown to control acute inflammation in rat SMG during retroductal viral delivery [18]. Furthermore, DEX serves as a potent anti-inflammatory drug that inhibits polymorphonuclear neutrophil function and tissue invasion during acute inflammatory responses [19]. Additionally, DEX causes a reduction of systemic cytokine expression in several conditions [20-22]. Corticosteroids, however, often result in debilitating side effects involving multiple organ systems [23], increasing the appeal of alternative anti-inflammatory therapies, such as resolvins.
In the present study, we compared the effects of AT-RvD1 and DEX on the inflammatory features (i.e., lymphocytic infiltration, cytokine expression and apoptosis) observed in SMG from the well-established NOD/ShiLtJ SS-like mouse model that exhibits many features of the human disease including lymphocytic infiltration and cytokine upregulation [24].
Materials and Methods
Experimental animals
Female NOD/ShiLtJ mice were treated via tail vein injection twice a week with NaCl (0.9%, used as negative control), different doses of the stable RvD1 analog, AT-RvD1 (0.01-0.1 mg/kg) and dexamethasone (DEX, 4.125-8.25 mg/Kg) from 4 weeks of age until 18 weeks of age (Figure 1). At 18 weeks of age the mice were anesthetized with 80-100 mg/kg Ketamine + 5 mg/kg Xylazine and subsequently euthanized by abdominal exsanguination. All animal usage, anesthesia, and surgeries were conducted under the strict guidelines and approval of the University of Utah Institutional Animal Care and Use Committee.
.
Figure 1: Diagram showing experimental treatments using AT-RvD1 and dexamethasone. 4 week-old female NOD/ShiLtJ mice were treated via tail vein injections with the following treatments: NaCl (0.9%), AT-RvD1 (0.01-0.1 mg/kg) and dexamethasone (DEX; 8.25 mg/Kg) 2 times a week for 14 weeks. At 18 weeks of age, submandibular glands were removed, frozen, and stained with Hematoxylin & Eosin, with sections visualized on a Leica DMI6000B Inverted Microscope.
View Figure 1
Histopathological evaluation
At 18 weeks of age, NOD/ShiLtJ mice were sacrificed and SMG were removed, frozen, sectioned, and stained with hematoxylin & eosin. Sections from the top, middle, and bottom of the gland were visualized using an Olympus BX53 upright research microscope (Olympus, Tokyo, Japan). Grading of SMG histological sections was performed as described by Chisholm & Mason [25]. Briefly, numerical scores were assigned as follows, 1: Slight infiltrate, 2: Moderate infiltrate of less than one focus per 4 mm2, 3: One focus per 4 mm2, 4: More than one focus per 4 mm2. Focus represents an aggregate of 50 or more lymphocytes.
RNA extraction and preparation of cDNA
Total RNA (from nine different frozen SMG, three from each of the following treatments: NaCl 0.9%, AT-RvD1 0.1 mg/kg and DEX 8.25 mg/kg) was extracted using the Bio-Rad Aurum Total RNA Mini Kit (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. RNA quantity was assessed using a BioTek Epoch microplate spectrophotometer and corresponding software Gen 5 Ver. 2.06 (BioTek, Winooski, VT). Each sample was diluted to 30 ng/μl in Elution solution (Bio-Rad). RNA purity was assessed by measurement of A260/A280 in a BioTek Epoch microplate spectrophotometer. RNA (0.5 μg) was then reverse transcribed to cDNA using the Bio-Rad iScript Advanced cDNA Synthesis Kit (Bio-Rad).
PCR array
Detection and quantification of mouse gene expression was performed with three different SMG per mouse treatment (i.e., NaCl 0.9%, AT-RvD1 0.1 mg/kg, DEX 8.25 mg/kg) using both the SS and the Apoptosis and Survival Tier 1 PrimePCR Pathway Plates, according to the manufacturer's instructions (Bio-Rad). These kits profile the expression of genes encoding proteins that are important in apoptosis, survival and SS. Quantitative PCR was performed for each sample individually using a CFX Connect Thermocycler (Bio-Rad). All results were normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). All replicates were then pooled to determine the relative expression level of each gene between the control and treatment groups. A high sensitivity genomic DNA (gDNA) control primer was used to detect non-transcribed gDNA contamination. CT values of gDNA were greater than 35 for all replicate PCR samples run, indicating that the level of gDNA contamination was minimal and did not affect gene expression profiling results. Data were analyzed using the CFX Manager software, version 3.1 (Bio-Rad). Statistical significance was determined using a t-test.
Western blot analysis
One half of a SMG was lysed in 200 μl of 2 × Laemmli buffer (120 mM Tris-HCl, pH 6.8, 10% (v/v) glycerol, 2% (w/v) SDS, 1 mM dithiothreitol and 0.002% (w/v) bromophenol blue), sonicated for 60 sec with a sonic dismembrator (model 120; microtip; amplification 45%; Fisher Scientific, Pittsburgh, PA), and boiled for 5 min. Twenty microliters of SMG lysates were subjected to electrophoresis with 4-15% (w/v) Mini-Protean TGX Precast Gels (Bio-Rad) and transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked for 1 h with 3% (w/v) bovine serum albumin (BSA) in Tris-buffered saline [0.137M NaCl, 0.025M Tris (hydroxymethyl)aminomethane, pH 7.4] containing 0.1% (v/v) Tween-20 (TBST), then immunoblotted overnight at 4°C in TBST containing 3% (w/v) BSA with rabbit anti-phospho Akt (1:500 dilution; Cell Signaling Technology). After incubation, membranes were washed three times for 20 min each with TBST and incubated for 1 h with peroxidase-linked goat anti-rabbit IgG antibody (1:2,000 dilution; Cell Signaling Technology). The membranes were treated with Clarity Western ECL Substrate (Bio-Rad), and protein bands were visualized and quantitated using a ChemiDoc MP and Image Lab v4.1 software (Bio-Rad). Membranes were treated with Restore Western Blot Stripping Buffer (Thermo Scientific, Rockford, IL) and re-probed with rabbit anti-pan Akt (1:500 dilution; Cell Signaling Technology) and with rabbit anti-pan-actin (1:1000 dilution; Cell Signaling Technology) for signal normalization followed by peroxidase-linked goat anti-rabbit IgG antibody (1:2,000 dilution; Cell Signaling Technology).
TUNEL analysis
Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assays were performed with a Click-iT TUNEL Alexa Fluor 488 Imaging Assay (Life Technologies, Grand Island, NY) following the manufacturer's instructions using TO-PRO-3 iodide (Life Technologies) as a nuclear marker. Briefly, frozen sections were fixed with 4% paraformaldehyde in PBS at room temperature for 15 min, permeabilized with Triton X-100 (1% in PBS) for 5 min and treated with 1% Sodium citrate for an additional 5 min. Then, sections were washed twice in PBS, and incubated with 100 μl of terminal deoxynucleotidyl transferase (TDT) reaction buffer (Component A) for 10 min at room temperature. The buffer was removed and the TUNEL reaction cocktail containing TDT was added, then sections were incubated in a humidified chamber overnight at room temperature. After treatment, sections were washed three times with 3% BSA in PBS for 5 min each and then incubated with 100 μl of Click-iT reaction mixture (containing Alexa Fluor 488 azide) for 1 h at room temperature, while being protected from light exposure. The sections were washed with 3% BSA in PBS. The staining was completed by incubation with TO-PRO-3 for 10 min at room temperature. The negative control was processed using a reaction mixture without terminal deoxynucleotidyl transferase (TDT) cocktail. The positive control section was incubated with DNase I (3 units/ml) for 1 h to induce DNA strand breaks. Positive and negative controls were performed in frozen mouse submandibular gland sections from C57BL/6 mice. TUNEL-positive nuclei images were taken using a Carl Zeiss LSM 710 confocal microscope, and visualized using the ZEN software (black edition; Carl Zeiss, Thornwood, NY). TUNEL-positive staining was calculated from a tile image of entire tissue sections, and quantified by mean intensity of images ( Supplemental Figure 1 and Supplementary Table 1).
Statistical analysis
Data presented are means ± S.E.M. of results from three or more determinations. Graphpad Prism software (Graphpad Software, Inc., La Jolla, CA) was used to perform one-way ANOVA, followed by pairwise post hoc multiple comparisons using Dunnett's test to generate P-values and 95% confidence intervals (CI) of the differences of the means. P-values equal to or less than 0.05 represent significant differences between experimental groups. A 4-fold regulation threshold was used to determine significant differences in gene microarray studies.
Results
Effects of AT-RvD1 and DEX on lymphocytic infiltration in NOD/ShiLtJ mouse SMG
The degree of lymphocytic infiltration was estimated by counting the number of lymphocytes in SMG from NOD/ShiLtJ mice as described in MATERIALS AND METHODS. As shown in table 1 and Supplemental Figure 2, SMG from NOD/ShiLtJ mice treated with NaCl (0.9%, used as negative controls) displayed a focus score of 4 in at least two out of three sections from each gland. Similarly, SMG from NOD/ShiLtJ mice treated with AT-RvD1 (0.1 mg/kg, used as target group) displayed focus scores comparable to SMG from mice treated with saline. In contrast, sections of SMG from mice treated with DEX (8.25 mg/kg) displayed a decrease in focus scores with the majority of the sections observed being 1 or 2. One gland displayed a focus score of 4 in two of the three sections. These results indicated that DEX treatment is effective in decreasing lymphocytic infiltration in these mice.
![]()
Table 1: Focus scores in mouse submandibular glands after NaCl, AT-RvD1, and dexamethasone treatments.
View Table 1
Effects of AT-RvD1 and DEX on inflammatory genes in mouse SMG
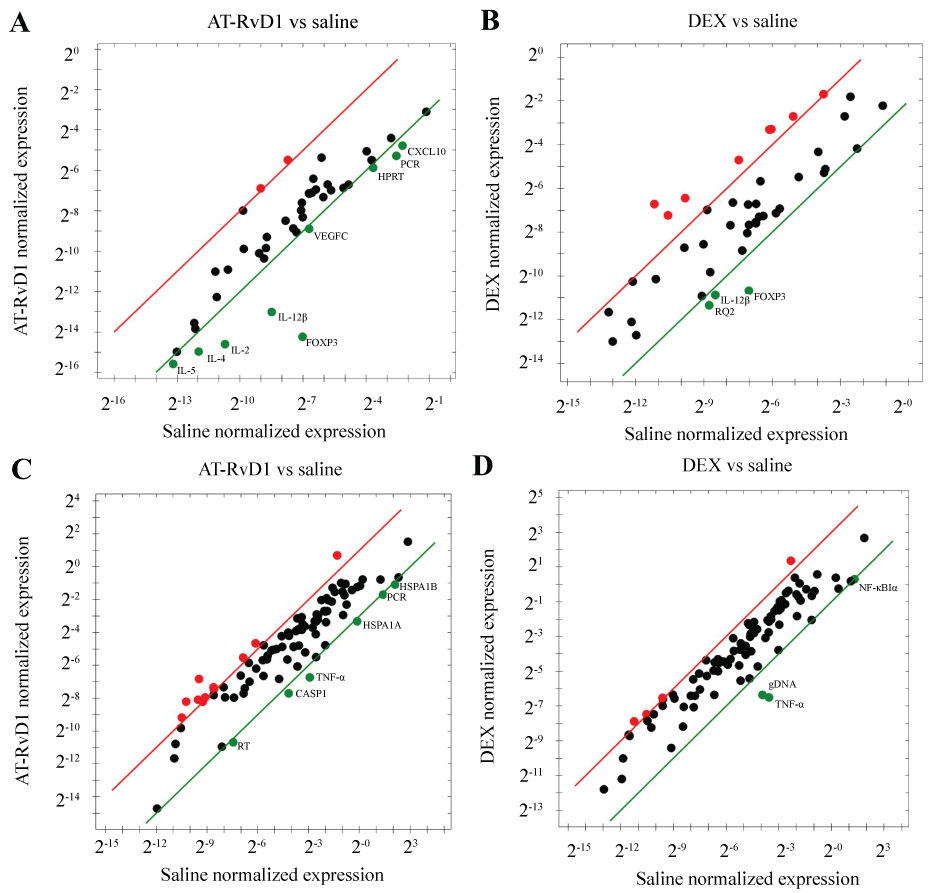
To determine whether AT-RvD1 and DEX treatment affected the expression of SS-associated inflammatory genes, we performed a microarray analysis. As shown in figure 2A, systemic AT-RvD1 (0.1 mg/kg) treatment caused a significant downregulation of the SS-associated inflammatory genes: CXCL10, IL-2, IL-4, IL-5, and IL-12β in SMG from NOD/ShiLtJ mice as compared to SMG from mice treated with saline. As shown in figure 2B, systemic DEX (8.25 mg/kg) treatment caused a significant downregulation of the SS-associated inflammatory genes: Foxp3 and IL-12β in SMG from NOD/ShiLtJ mice as compared to SMG from mice treated with saline.
.
Figure 2: Detection and quantification of mouse apoptosis and survival gene expression and SS gene expression. Expression levels of genes important to apoptosis and survival as well as Sjögren's syndrome (SS) were detected and quantified by PCR array as described in MATERIALS AND METHODS and represented on a scatter plot. Black dots indicate no change in gene expression from control, red dots indicate upregulation, and green dots indicate downregulation. Regression lines for upregulation (red) and downregulation (green) are also shown. (A) Expression levels after treatment of AT-RvD1 (0.1 mg/kg) as compared to control treatment of NaCl (0.9%) using a SS gene array; (B) Expression levels after dexamethasone (DEX) treatment (8.25 mg/kg) as compared to control treatment of NaCl (0.9%) using a SS gene array; (C) Expression levels after treatment of At-RvD1 (0.1 mg/kg) as compared to control treatment of NaCl using an apoptotic and survival gene array; (D) Expression levels after dexamethasone (DEX) treatment (8.25 mg/kg) as compared to control treatment of NaCl (0.9%) using an apoptotic and survival gene array.
View Figure 2
To determine whether AT-RvD1 and DEX treatment affected the expression of apoptotic and survival genes, we performed a second microarray analysis. As shown in figure 2C, systemic AT-RvD1 (0.1 mg/kg) treatment caused a significant downregulation (4-fold regulation threshold) of the pro-apoptotic genes: TNF-α, Casp1, Hspa1a, and Hspa1b in SMG from NOD/ShiLtJ mice as compared to SMG from mice treated with saline. As shown in figure 2D, systemic DEX (8.25 mg/kg) treatment caused a significant downregulation of the pro-apoptotic genes: Foxp3, IL-12β, TNF-α and NF-κB in SMG from NOD/ShiLtJ mice as compared to SMG from mice treated with saline.
Effects of AT-RvD1 and DEX on cell death in mouse SMG
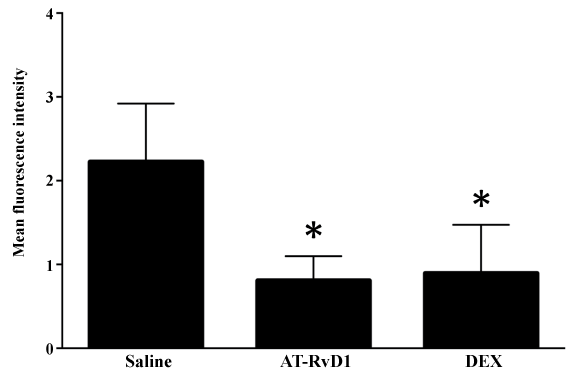
To assess the effect of AT-RvD1 and DEX treatment on cell death in SMG of NOD/ShiLtJ mice, we quantified the mean intensity of TUNEL positive cells. As shown in figure 3, SMG from NOD/ShiLtJ mice systemically treated with AT-RvD1 (0.1 mg/kg) showed a significant decrease in mean intensity of TUNEL positive cells as compared to SMG from mice treated with saline. Similarly, SMG from mice systemically treated with DEX (8.25 mg/kg) showed a significant decrease in mean intensity of TUNEL positive cells as compared to SMG from mice treated with saline. These results indicate that both AT-RvD1 and DEX were effective in decreasing the overall apoptosis levels in SMG from NOD/ShiLtJ mice.
.
Figure 3: Aspirin-triggered RvD1 (AT-RvD1) significantly decreases the level of apoptotic cell death in SMG from NOD/ShiLtJ mice. NOD/ShiLtJ mice were systemically injected via tail vein injections with the following treatments: (A) NaCl (0.9%), (B) AT-RvD1 (0.01-0.1 mg/kg) and (C) dexamethasone (DEX; 8.25 mg/Kg) 2 times a week for 14 weeks. At 18 weeks of age, submandibular glands were removed, frozen, and sections from were fixed and evaluated by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL). The results are from 3 biological replicates, where * P ≤ 0.05 indicates a significant difference from saline controls.
View Figure 3
Effects of AT-RvD1 and DEX on phosphorylation of Akt in mouse SMG
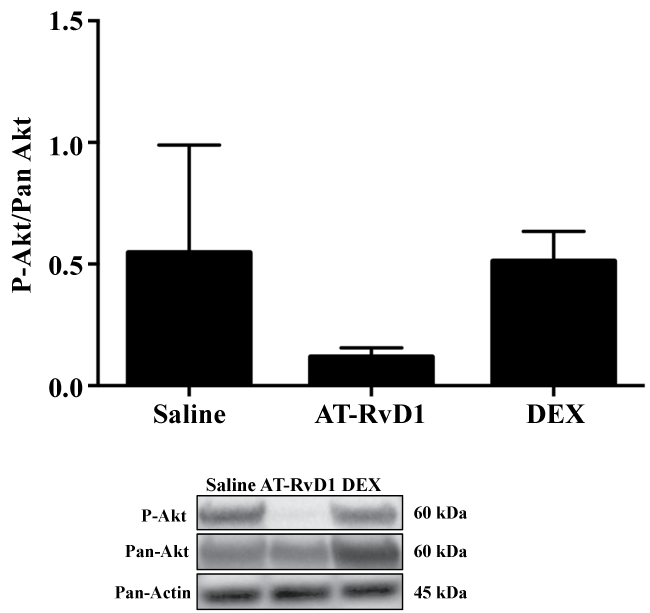
Our previous studies showed that RvD1 is able to cause phosphorylation of the survival Akt pathway, blocking apoptosis in salivary cells [13]. Therefore, to determine whether decreased apoptosis correlated with Akt phosphorylation, we decided to test whether SMG from NOD/ShiLtJ mice systemically treated with AT-RvD1 displayed a sustained activation of this pathway. As shown in figure 4, systemic treatment with AT-RvD1 (0.1 mg/kg) did not cause a sustained phosphorylation of Akt in SMG from NOD/ShiLtJ mice as compared to SMG from NOD/ShiLtJ mice treated with saline.
.
Figure 4: Aspirin-triggered RvD1 (AT-RvD1) does not cause sustained Akt phosphorylation in NOD/ShiLtJ mice. NOD/ShiLtJ mice were systemically injected via tail vein injections with the following treatments: (A) NaCl (0.9%), (B) AT-RvD1 (0.01-0.1 mg/kg) and (C) dexamethasone (DEX; 8.25 mg/Kg) 2 times a week for 14 weeks. At 18 weeks of age, submandibular glands were removed and SMG freshly isolated cells were lysed in Laemmli buffer, subjected to SDS-PAGE, and probed with primary antibody rabbit anti-phospho Akt.
View Figure 4
Discussion
Salivary gland inflammation and dysfunction are hallmarks of SS [26]. Current anti-inflammatory treatments do not fully relieve salivary gland chronic inflammation without causing adverse side effects [6,16]. The complex interaction between multiple inflammatory pathways requires future studies to achieve a deeper mechanistic understanding of SS etiology and pathogenesis. Such complex interactions suggest that a combinatorial treatment may be a better therapeutic approach than individual treatments. Due to its lack of adverse side effects, AT-RvD1 offers an attractive alternative option to the use of corticosteroids. RvD1 has shown to be useful in treating a variety of conditions such as experimental colitis, alveolar epithelial cell injury, smoke-induced lung inflammation, colon cancer, rheumatoid arthritis, chronic obstructive pulmonary disease, ischemic injuries and uveitis [27-34]. Moreover, our previous in vitro studies indicate that RvD1 and AT-RvD1 block inflammatory signals in the rat parotid Par-C10 cell line and in freshly isolated submandibular gland cells [13,14], suggesting a potential therapeutic benefit for SS in vivo.
Our results indicate that AT-RvD1 treatment was not effective in decreasing lymphocytic infiltration in NOD/ShiLtJ mice (Table 1). In contrast, systemic treatment with DEX prevented lymphocytic infiltration in two out of three NOD/ShiLtJ mice (Table 1). Reduction of lymphocytic infiltration has been observed previously with DEX treatment for patients suffering from autoimmune diseases, such as rheumatoid arthritis [35] and systemic lupus erythematous [36]. While DEX appeared to have a more directly observable effect on lymphocytic infiltration than AT-RVD1 as evidenced by histopathological evaluation, we wanted to determine the effects of both treatments on other indicators of inflammation. To this end, we performed gene expression analyses using SS- and apoptosis-associated gene expression panels. We found that AT-RVD1 and DEX affected the expression of mostly non-overlapping groups of genes (Figure 2A, Figure 2B, Figure 2C and Figure 2D). AT-RvD1 significantly reduced the expression of SS-related pro-inflammatory cytokines as well as apoptotic and survival genes (e.g., IL-2, -4, -5, and -12β, TNF-α, Casp1, Foxp3, and Cxcl10, Figure 2A) in SMG from NOD/ShiLtJ mice as opposed to mice treated with saline (used as negative control). Interestingly, DEX was able to downregulate IL-12β (similar to AT-RvD1); however, it significantly downregulated different pro-inflammatory genes compared to those downregulated by AT-RvD1 (Figure 2B). Our results are consistent with previous findings showing that RvD1-reduces cytokine expression in alveolar epithelium [29], primary human lung fibroblasts [30], colonic epithelium [31], and in rat eyes [33]. In the case of DEX, our results are also consistent with previous studies showing reduction of cytokine expression in human corneal epithelium [37], airway epithelium [38] and in brain microvascular endothelial cells [39]. The different effects elicited by AT-RvD1 and DEX on anti-inflammatory cytokine gene expression suggest that using them in combination may be an effective treatment for SS.
Of particular interest is gene expression for the pro-inflammatory cytokine, TNF-α, which was downregulated in both AT-RvD1 and DEX treatments. TNF-α is secreted by macrophages, mature T helper cells and epithelial cells, and has been identified as a key regulator of inflammatory responses [40]. The role of TNF-α in the pathogenesis of primary SS has not been fully evaluated; however, there are a few studies indicating that TNF-α may play a role [2,4,40]. Specifically, TNF-α has been shown to be upregulated along with its receptors in serum [4] and salivary glands [2] from SS patients. Neutralization of TNF-α in NOD mice by transgenic expression of soluble TNF-receptor significantly reduced lymphocytic infiltration in SMG and lacrimal glands associated with decreased expression of the cell adhesion molecules, vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1) [41]. These results suggest a role for TNF-α in the initiation and progression of salivary and lacrimal gland destruction in NOD mice. Our previous studies indicate that TNF-α upregulates cell adhesion molecules and binding of lymphocytes in the human submandibular gland cell line (HSG) [42]. Additionally, TNF-α causes salivary gland dysfunction of Par-C10 cells and primary cells [13,14,43,44]. Interestingly, blocking TNF-α alone did not show improvement in patients using anti-TNF-α therapy [45], suggesting that other cytokines may also contribute to SS pathogenesis. This pilot study will serve as a foundation for our future pre-clinical study in which we will increase the population size and study whether other cytokines (such as IFN-γ and IL-10) are downregulated after AT-RvD1 treatment.
Our findings suggest that AT-RvD1 and DEX individually affect apoptosis independently from lymphocytic infiltration (Figure 3). These results are consistent with previous in vivo studies showing that RvD1 reduces apoptosis induced by lipopolysaccharide (LPS) in the liver [46]. Additionally, RvD1 reduced LPS-induced pulmonary cellular apoptosis in mice [47]. Furthermore, AT-RvD1 treatment significantly decreased apoptosis and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling in acute lung injury [48].
Similarly, the use of DEX has been associated with decreased apoptosis but with deleterious effects including a reduction in insulin-secreting cells [49], a decrease of neural progenitor cells in the hippocampus [50] and diminishing oligodendrocyte progenitor cells resulting in hypomyelination. More recently, a study showed that DEX increases apoptosis in osteoblast and osteocytes, leading to bone loss [51]. These results highlight the positive impact of AT-RvD1 and the secondary effects that could be caused by DEX and indicate the need for further studies combining these two treatments to resolve inflammation associated with SS.
Our studies showed that neither AT-RvD1 nor DEX caused a sustained phosphorylation of Akt in mouse SMG (Figure 4). Furthermore, Akt phosphorylation did not correlate with the decreased apoptosis observed in mice treated under these conditions (Figure 4). Our previous studies showed that activation of ALX/FPR2 with AT-RvD1 induces transient Akt phosphorylation in rat parotid Par-C10 cells [13] as well as in mouse SMG [14]. These results indicate that AT-RvD1 initiates rapid signaling mechanisms to block stress generated by tissue culture conditions. Therefore, it is possible that Akt displayed a transient unmeasurable phosphorylation at the time point studied (at least in the case of AT-RvD1). In contrast, DEX has been shown to inhibit Akt phosphorylation in several tissues caused by dexamethasone-induced reductions in PI3 kinase activity. Future studies to measure Akt phosphorylation over time will be necessary to better understand the AT-RvD1 and DEX mechanisms to prevent cell apoptosis [52].
We were able to test the feasibility of using AT-RvD1 and DEX before using these treatments in a large scale population. In this pilot study, were able to examine effects and associations that warrant further studies (see review, [53]). Given our findings, future studies will determine whether AT-RvD1 and DEX alone and in combination prove to be useful in the treatment of experimental SS.
Acknowledgements
This work was supported by the NIH-NIDCR grants R01DE021697 and R01DE022971 (to OJB).
References
-
Luciano N, Valentini V, Calabrò A, Elefante E, Vitale A, et al. (2015) One year in review 2015: Sjögren's syndrome. Clin Exp Rheumatol 33: 259-271.
-
Fox RI, Kang HI, Ando D, Abrams J, Pisa E (1994) Cytokine mRNA expression in salivary gland biopsies of Sjogren's syndrome. J Immunol 152: 5532-5539.
-
Delaleu N, Nguyen CQ, Peck AB, Jonsson R (2011) Sjogren's syndrome: studying the disease in mice. Arthritis Res Ther 13: 217.
-
Garcíc-Carrasco M, Font J, Filella X, Cervera R, Ramos-Casals M, et al. (2001) Circulating levels of Th1/Th2 cytokines in patients with primary Sjögren's syndrome: correlation with clinical and immunological features. Clin Exp Rheumatol 19: 411-415.
-
Roescher N, Tak PP, Illei GG (2009) Cytokines in Sjögren's syndrome. Oral Dis 15: 519-526.
-
Ramos-Casals M, Tzioufas AG, Stone JH, Sisó A, Bosch X (2010) Treatment of primary Sjögren syndrome: a systematic review. JAMA 304: 452-460.
-
Orr SK, Colas RA, Dalli J, Chiang N, Serhan CN (2015) Proresolving actions of a new resolvin D1 analog mimetic qualifies as an immunoresolvent. Am J Physiol Lung Cell Mol Physiol 308: L904-911.
-
Serhan CN (2014) Pro-resolving lipid mediators are leads for resolution physiology. Nature 510: 92-101.
-
Serhan CN, Chiang N, Van Dyke TE (2008) Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8: 349-361.
-
Dalli J, Ramon S, Norris PC, Colas RA, Serhan CN (2015) Novel proresolving and tissue-regenerative resolvin and protectin sulfido-conjugated pathways. FASEB J 29: 2120-2136.
-
Serhan CN, Dalli J, Karamnov S, Choi A, Park CK, et al. (2012) Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J 26: 1755-1765.
-
Van Dyke TE, Hasturk H, Kantarci A, Freire MO, Nguyen D, et al. (2015) Proresolving nanomedicines activate bone regeneration in periodontitis. J Dent Res 94: 148-156.
-
Odusanwo O, Chinthamani S, McCall A, Duffey ME, Baker OJ (2012) Resolvin D1 prevents TNF-a-mediated disruption of salivary epithelial formation. Am J Physiol Cell Physiol 302: 1331-1345.
-
Nelson JW, Leigh NJ, Mellas RE, McCall AD, Aguirre A, et al. (2014) ALX/FPR2 receptor for RvD1 is expressed and functional in salivary glands. Am J Physiol Cell Physiol 306: C178-185.
-
Leigh NJ, Nelson JW, Mellas RE, Aguirre A, Baker OJ (2014) Expression of resolvin D1 biosynthetic pathways in salivary epithelium. J Dent Res 93: 300-305.
-
Ramos-Casals M, Tzioufas AG, Font J (2005) Primary Sjögren's syndrome: new clinical and therapeutic concepts. Ann Rheum Dis 64: 347-354.
-
Izumi M, Eguchi K, Nakamura H, Takagi Y, Kawabe Y, et al. (1998) Corticosteroid irrigation of parotid gland for treatment of xerostomia in patients with Sjögren's syndrome. Ann Rheum Dis 57: 464-469.
-
Adesanya MR, Redman RS, Baum BJ, O'Connell BC (1996) Immediate inflammatory responses to adenovirus-mediated gene transfer in rat salivary glands. Hum Gene Ther 7: 1085-1093.
-
Perretti M, Chiang N, La M, Fierro IM, Marullo S, et al. (2002) Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med 8: 1296-1302.
-
El Azab SR, Rosseel PM, de Lange JJ, Groeneveld AB, van Strik R, et al. (2002) Dexamethasone decreases the pro- to anti-inflammatory cytokine ratio during cardiac surgery. Br J Anaesth 88: 496-501.
-
Remmelts HH, Meijvis SC, Biesma DH, van Velzen-Blad H, Voorn GP, et al. (2012) Dexamethasone downregulates the systemic cytokine response in patients with community-acquired pneumonia. Clin Vaccine Immunol 19: 1532-1538.
-
Schuld A, Kraus T, Haack M, Hinze-Selch D, Zobel AW, et al. (2001) Effects of dexamethasone on cytokine plasma levels and white blood cell counts in depressed patients. Psychoneuroendocrinology 26: 65-76.
-
Schäcke H, Döcke WD, Asadullah K (2002) Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther 96: 23-43.
-
Nguyen CQ, Cha SR, Peck AB (2007) Sjögren's syndrome (SjS)-like disease of mice: the importance of B lymphocytes and autoantibodies. Front Biosci 12: 1767-1789.
-
Chisholm DM, Mason DK (1968) Labial salivary gland biopsy in Sjögren's disease. J Clin Pathol 21: 656-660.
-
Jonsson R, Brun JG (2010) Sjögren's syndrome. 2010: Wiley Online Library.
-
Bento AF1, Claudino RF, Dutra RC, Marcon R, Calixto JB (2011) Omega-3 fatty acid-derived mediators 17 (R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. J Immunol 187: 1957-1969.
-
Bozinovski S, Anthony D, Anderson GP, Irving LB, Levy BD, et al. (2013) Treating neutrophilic inflammation in COPD by targeting ALX/FPR2 resolution pathways. Pharmacol Ther 140: 280-289.
-
Cox R, Phillips O, Arias S, Mandry M, Fukumoto J, et al. (2015) Aspirin-Triggered Resolvin D1 Protects Against Cytokine Induced Alveolar Epithelial Cell Injury. The FASEB Journal, 2015. 29: 863.18.
-
Hsiao HM, Sapinoro RE, Thatcher TH, Croasdell A, Levy EP, et al. (2013) A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLoS One 8: e58258.
-
Janakiram NB, Rao CV (2009) Role of lipoxins and resolvins as anti-inflammatory and proresolving mediators in colon cancer. Curr Mol Med 9: 565-579.
-
Norling LV, Perretti M (2013) The role of omega-3 derived resolvins in arthritis. Curr Opin Pharmacol 13: 476-481.
-
Settimio R, Clara DF, Franca F, Francesca S, Michele D (2012) Resolvin D1 reduces the immunoinflammatory response of the rat eye following uveitis. Mediators Inflamm 2012: 318621.
-
Zhang T, Shu HH, Chang L, Ye F, Xu KQ, et al. (2015) Resolvin D1 protects against hepatic ischemia/reperfusion injury in rats. Int Immunopharmacol 28: 322-327.
-
De A, Blotta HM, Mamoni RL, Louzada P, Bertolo MB, et al. (2002) Effects of dexamethasone on lymphocyte proliferation and cytokine production in rheumatoid arthritis. J Rheumatol 29: 46-51.
-
Ho CY, Wong CK, Li EK, Lam WK (2001) Effects of dexamethasone on the expression of Fas molecules and apoptosis of lymphocytes in patients with systemic lupus erythematosus. Immunol Invest 30: 231-243.
-
Djalilian AR, Nagineni CN, Mahesh SP, Smith JA, Nussenblatt RB, et al. (2006) Inhibition of inflammatory cytokine production in human corneal cells by dexamethasone, but not cyclosporin. Cornea 25: 709-714.
-
van Wetering S, Mannesse-Lazeroms SP, van Sterkenburg MA, Hiemstra PS (2002) Neutrophil defensins stimulate the release of cytokines by airway epithelial cells: modulation by dexamethasone. Inflamm Res 51: 8-15.
-
Blecharz KG, Haghikia A, Stasiolek M, Kruse N, Drenckhahn D, et al. (2010) Glucocorticoid effects on endothelial barrier function in the murine brain endothelial cell line cEND incubated with sera from patients with multiple sclerosis. Mult Scler 16: 293-302.
-
Bradley JR (2008) TNF-mediated inflammatory disease. J Pathol 214: 149-160.
-
Hunger RE, Müller S, Laissue JA, Hess MW, Carnaud C, et al. (1996) Inhibition of submandibular and lacrimal gland infiltration in nonobese diabetic mice by transgenic expression of soluble TNF-receptor p55. J Clin Invest 98: 954-961.
-
Baker OJ, Camden JM, Rome DE, Seye CI, Weisman GA (2008) P2Y2 nucleotide receptor activation up-regulates vascular cell adhesion molecule-1 [corrected] expression and enhances lymphocyte adherence to a human submandibular gland cell line. Mol Immunol 45: 65-75.
-
Baker OJ, Camden JM, Redman RS, Jones JE, Seye CI, et al. (2008) Proinflammatory cytokines tumor necrosis factor-alpha and interferon-gamma alter tight junction structure and function in the rat parotid gland Par-C10 cell line. Am J Physiol Cell Physiol 295: 1191-201.
-
Baker OJ, Schulz DJ, Camden JM, Liao Z, Peterson TS, et al. (2010) Rat parotid gland cell differentiation in three-dimensional culture. Tissue Engineering Part C: Methods 16: 1135-1144.
-
Mariette X, Ravaud P, Steinfeld S, Baron G, Goetz J, et al. (2004) Inefficacy of infliximab in primary Sjogren's syndrome: results of the randomized, controlled Trial of Remicade in Primary Sjogren's Syndrome (TRIPSS). Arthritis Rheum 50: 1270-1276.
-
Murakami T, Suzuki K, Tamura H, Nagaoka I (2011) Suppressive action of resolvin D1 on the production and release of septic mediators in D-galactosamine-sensitized endotoxin shock mice. Exp Ther Med 2: 57-61.
-
Xie W, Wang H, Wang L, Yao C, Yuan R, et al. (2013) Resolvin D1 reduces deterioration of tight junction proteins by upregulating HO-1 in LPS-induced mice. Lab Invest 93: 991-1000.
-
Cox R Jr, Phillips O, Fukumoto J, Fukumoto I, et al. (2015) Enhanced Resolution of Hyperoxic Acute Lung Injury as a result of Aspirin Triggered Resolvin D1 Treatment. Am J Respir Cell Mol Biol 53: 422-435.
-
Ranta F, Avram D, Berchtold S, Düfer M, Drews G, et al. (2006) Dexamethasone induces cell death in insulin-secreting cells, an effect reversed by exendin-4. Diabetes 55: 1380-1390.
-
Sze CI, Lin YC, Lin YJ, Hsieh TH, Kuo YM, et al. (2013) The role of glucocorticoid receptors in dexamethasone-induced apoptosis of neuroprogenitor cells in the hippocampus of rat pups. Mediators Inflamm 2013: 628094.
-
Sato AY, Tu X, McAndrews KA, Plotkin LI, Bellido T (2015) Prevention of glucocorticoid induced-apoptosis of osteoblasts and osteocytes by protecting against endoplasmic reticulum (ER) stress in vitro and in vivo in female mice. Bone 73: 60-68.
-
Yu-Shengyou1, Li Y (2013) Dexamethasone inhibits podocyte apoptosis by stabilizing the PI3K/Akt signal pathway. Biomed Res Int 2013: 326986.
-
Thabane L, Ma J, Chu R, Cheng J, Ismaila A, et al. (2010) A tutorial on pilot studies: the what, why and how. BMC Med Res Methodol 10: 1.
