

Journal of Genetics and Genome Research
Genomic and Genotyping Characterization of Haplotype-Based Polymorphic Microsatellites in Prunus
Chunxian Chen*, Clive H. Bock, Tom G. Beckman, Bruce W. Wood and William R. Okie
United States Department of Agriculture, Agricultural Research Service, Southeastern Fruit and Nut Tree Research Lab, USA
*Corresponding author:
C. Chen, United States Department of Agriculture, Agricultural Research Service, Southeastern Fruit and Nut Tree Research Lab, 21 Dunbar Road, Byron, GA 31008, USA, Tel: 478-956-6467,Fax: 478-956-2929, E-mail: chunxian.chen@ars.usda.gov
J Genet Genome Res, JGGR-2-014, (Volume 2, Issue 1), Research Article; ISSN: 2378-3648
Received: April 14, 2015 | Accepted: May 27, 2015 | Published: May 30, 2015
Citation: Chen C, Bock CH, Beckman TG, Wood BW, Okie WR (2015) Genomic and Genotyping Characterization of Haplotype-Based Polymorphic Microsatellites in Prunus. J Genet Genome Res 2:014. 10.23937/2378-3648/1410014
Copyright: © 2015 Chen C, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Efficient utilization of microsatellites in genetic studies remains impeded largely due to the unknown status of their primer reliability, chromosomal location, and allele polymorphism. Discovery and characterization of microsatellite polymorphisms in a taxon will disclose the unknowns and gain new insights into the polymorphic alleles. In this study, we revealed the polymorphism status, primer categorization, chromosomal distribution, gene function, and genotyping performance of 319 haplotype-based polymorphic microsatellites (HPM) in expressed sequence tags (EST) of Prunus species, including peach, apricot, almond, plums, and cherries. Of the HPM, 262 are between two EST haplotypes and 57 are among three and more EST. In terms of species, 127 microsatellite polymorphisms are from different EST of peach, 108 from different EST between peach and other species, and 84fromdifferent EST between non-peach species. Based on the primer sequence alignments on the peach genome, there was one HPM per 678 kb and the 319 HPM were grouped into seven categories. The primers from the "deletion" category tended to yield higher allele numbers and polymorphism information content (PIC) values. Statistical analysis revealed the mean allele number, heterozygosity, PIC, and gene diversity value were all significantly higher in the HPM than in the haplotype-based non-polymorphic microsatellites (HNM), suggesting utilization of HPM markers could substantially increase the likelihood of allele polymorphism. Of the 234 unigenes annotated, 99 (42.3%) were categorized into binding function and 84 (35.9%) into catalytic activity, implying that these polymorphic alleles might have evolved primarily to play regulatory roles or catalyze enzymatic reactions.
Keywords
Simple sequence repeat, Co-dominance, Allelic variation, Functional diversity
Introduction
Microsatellite (also called simple sequence repeat - SSR) is one of the most widely used co-dominant marker types, because this type of markers are relatively abundant in genomes, easy to develop through sequence mining, transferable among related genotypes, and moderate in genotyping cost and throughput [1-4]. Computational microsatellite and single nucleotide polymorphism (SNP) discovery has become relatively easy as expressed sequence tag (EST) and genome sequences are exponentially growing in many crops [5,6]. Polymorphisms at a locus are derived from nucleotide variations in the expressed or genomic haplotypes (alleles) of the locus. Unlike a SNP, in which the transition or transversion polymorphism is computationally validated by alignment of redundant EST haplotypes or genomic reads [7,8], a microsatellite motif and its flanking forward and reverse primer are typically derived from an individual EST or genomic sequence, or from an unigene or genomic contig (consensus sequence generated by assembler), and no polymorphism is computationally predicted in its microsatellite-included amplicon [5]. Microsatellite polymorphisms usually result from varying numbers of repeat units in a length of one to six nucleotides. Unfortunately, the amplicon polymorphism status and performance reliability of a microsatellite primer are unknown until it is genotyped, so is its chromosomal location until the primer sequence is aligned to a reference genome. Consequently, high failure and low polymorphism rates and uneven chromosomal distribution are usually common in randomly selected microsatellite markers [3], which are the main drawbacks in utilization of microsatellite markers. Given microsatellite primer sequences aligned onto a reference genome, the performance reliability of optimally selected primers can be substantially improved, so can the chromosomal location of these primers be predicted [9]. Likewise, identification of haplotype-based polymorphic microsatellites (HPM) can greatly improve the real polymorphism rate of microsatellite markers in genotyping, as recently reported in several efforts to identify and utilize HPM, for example, in human [10], pepper [11], grapevine [12], and freshwater prawn [13]. However, partly due to relatively low frequency of HPM and highly variable numbers of repeat units, programs used for and species with computational identification of true HPM have been very limited [11,14], compared to SNP mining [4,7,8]. Development, characterization, and utilization of microsatellite markers have been reported in peach and other Prunus species, including early isolation from genomic and cDNA libraries and/or characterization of transportability across Prunus species [15-20], development from amplified fragment length polymorphism fragments [21], phylogenetic studies on peach genotypes [1,2], genetic mapping [22-25], and computational mining in Prunus ESTs [26]. But it appears no report involved particularly in computational mining and characterization of HPM in EST haplotypes of Prunus species or in polymorphism rate comparison between HPM and haplotype-based non-polymorphic microsatellites (HNM) in peach, although predictably computational discovery of HPM in Prunus may yield improved polymorphisms in these microsatellite markers, as reported in other studies [10,11].
The rapid innovation and reduced cost of next-generation sequencing technologies are driving the development and utilization of large-capacity SNP arrays for peach and other Prunus species [6,8,27,28], or other high-throughput assays, such as genotyping by sequencing [29]. However, along with SNP discovery, SNP genotyping on arrays or by sequencing generally is performed at high throughput and requires expensive proprietary instruments and computational capacities [28,30], which appears not economically practical for most routine, budget-constrained marker studies. Microsatellite genotyping, flexible in throughput and affordable in detection, remains more widely used, as demonstrated in many reports [1-3,22-25]. In a comparison of simulated datasets, the information content of microsatellite markers spacing at 7.5cM is slightly higher than that of SNP markers spacing at 3cM. At the map densities, microsatellites are found to be uniformly more informative than SNPs irrespective of their level of heterozygosity [31]. Another advantage of gene-based microsatellites results from an intriguing fact - substantially more microsatellites are found in the low-copy transcribed regions than in other regions of plant genomes [32].
In this study, HPM in Prunus ESTs will be mined out and characterized to gain new insights into genomic characterizations of these polymorphic alleles in the taxon, including the number of unigenes containing HPM, the annotated function of these unigenes, the status of polymorphisms, and the predicted categorization, performance reliability, and chromosomal location of primers. Selected primers will be used to compare the genotyping polymorphism rates between HPM and HNM.
Materials and Methods
Prunus ESTs and accession ID modifications
All ESTs from different Prunus species were summarized in Table 1. The long GenBank FASTA header of each sequence was simplified to its accession ID that was prefixed with a unique 2-letter abbreviation prior to assembly. Abbreviations were derived from the first two letters of the species name or the first letter plus another distinguish in letter if the first two letters were shared among species names, with one exception for the hybrid ("hy") (Table 1). For example, the abbreviation "pe" isused for Prunus persica, "so" for P. serotina, and "su" for P. serrulata. These prefixes were used to track the source species of ESTs in contigs and in the subsequent microsatellite mining and data analysis. After completion of the assembly process, each prefixed ID in the contigs was then suffixed with its belonging contig number to track its contig source.
![]()
Table 1: Prunus species with ESTs and prefixes used for EST source tracking
View Table 1
Mining HPM
A haplotype-based polymorphic microsatellite mining procedure was used in this study (Figure 1). In this mining process the species-prefixed accession ID of each individual EST in a contig was then suffixed with its contig number prior to microsatellite motif identification using MISA [5] and subsequent primer design using Primer3 [33] to track the species and contig source of ESTs when polymorphic microsatellite motifs were found. In the parameter file used by MISA was set 2-6, 3-4, 4-3, 5-3, and 6-3 (repeat unit length - minimal repeat number), and 100bp (the interval of adjacent microsatellites scored as the compound type), respectively. So the microsatellite motif types were designated p2, p3, p4, p5, or p6, defined by the repeat unit length of two, three, four, five or six nucleotides, in addition to the compound type. The primer results were saved in a tab-delimited text file and converted into an EXCEL file, to distinguish contigs with HPM from those with HNM among their ESTs. A contig would be categorized either as HNM if all ESTs in the contig contained the same motifs with the same number of repeats and at least one identical primer sequence (forward or reverse), or as HPM if at least two polymorphic microsatellites (with the same motif unit but different numbers of repeats) were found among some of its ESTs. To reduce redundancy in HPM ESTs, redundant ESTs from peach were always eliminated firstly if there were ESTs from other Prunus species that had the same polymorphic microsatellite motifs. After elimination of all redundant ESTs ultimately only one EST was kept to represent each unique allelic polymorphic microsatellite motif with a HPM contig. Secondly, those microsatellites with different motifs and primer sequences, which apparently belonged to different microsatellite loci within the same contigs, were excluded. Finally, a HPM within a contig contained ESTs sharing the same forward and/or reverse primer sequence flanking identical or variant motif units but with different numbers of repeat units for the motif. The variant motif units could be derived from nucleotide complementarity or discrepancies, and these HPM were categorized into "variant type" of motif units. For example, a motif was scored as [CAT] 4 in AGCATCATCATCATC, but its variant could be scored as [ATC] 5 in ATCATCATCATCATC because of the underlined G-T transversion, or [GAT] 4 or [GAT] 5 in ESTs complimentary to the two sequences, respectively. All these HPM are listed in the electronic supplementary material (ESM Table 1).
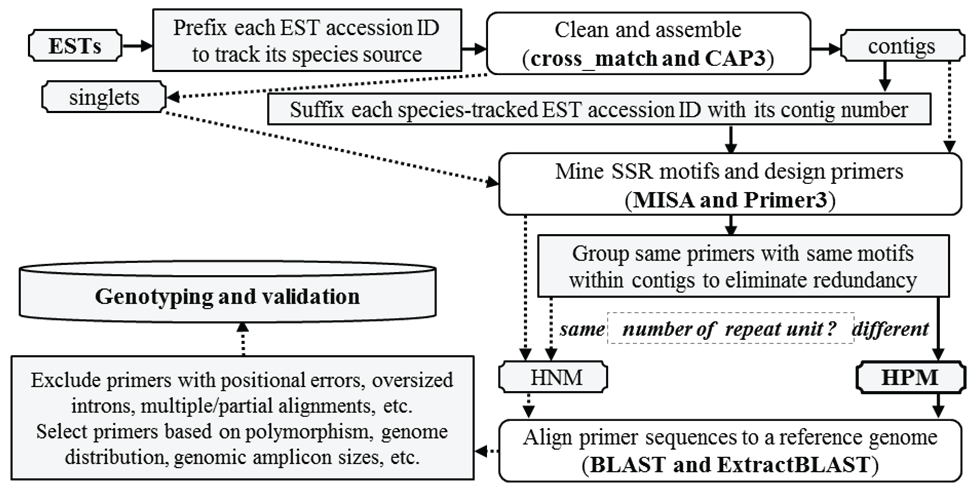
Figure 1: Flowchart showing the process for mining expressed haplotype-based polymorphic and non-polymorphic microsatellites (HPM and HNM). The bioinformatics programs in the parenthesis were used in the pipeline, including cross match [38] to remove vector DNA from ESTs, CAP3 [39] to assemble ESTs into unigenes, MISA [5] to find microsatellite motifs in ESTs, Primer3 [33] to design microsatellite-flanking primers, BLAST [40] to align primer sequences to the peach reference genome [34], and Extract BLAST (an in-house Java program) to parse BLAST output files into a tab text file and generate summaries. Non-polymorphic primers included those from singlet's and contigs with the same number of microsatellite unit among their ESTs, which were connected by dot arrows and not reported in this study. Polymorphic primers refer to those from contig with different numbers of SSR unit among their representative EST haplotypes.
View Figure 1
These HPM microsatellite primer sequences were formatted into the FASTA format for BLASTN at a cut-off e-value of 9e-03 (0.009) against the peach reference genome sequence [34]. The genomic amplicon size (GAS) of each primer was calculated by the subtraction between the maximal and the minimal value representing the four start and end alignment positions of the forward (F) and reverse (R) primer on the reference genome. Likewise, the amplicon size difference (ASD) of each aligned F and R primer was calculated by subtraction of the predicted EST amplicon size (EAS) from GAS. An ASD was used to categorize the primer into "no hit found" (NHF) if the F or R primer or both had no hit; "deletion" if the ASD was negative; "same size" if it was zero; "insertion" if it was positive and smaller than 20bp, a presumed minimum intron length cut-off; "intron (GAS<=500)" if it was over the cut-off and the intron-containing genomic amplicon was <=500bp; "intron (GAS>500)" if the intron-containing genomic amplicon was >500bp; or "error" if the ASD was too big to be a possible amplicon or intron, as described previously in detail. Randomly selected only from the "deletion", "same size", "insertion", and "intron (GAS<=500)" categories with a consideration on their relatively even distribution on 8 scaffolds of the peach reference genome, a subset of 96 HPM primers ((ESM Table 1, 2), along with a subset of 96 HNM primers (ESM Table 2), were used for comparison of the genotyping results.
Genotyping for polymorphism rate comparison between HPM and HNM primers
Genomic DNA extraction and microsatellite genotyping were performed as previously described [3] on four peach cultivars of different characteristics, 'Chinese Cling', 'Blazeprince', 'Helen Borchers' and 'Heath Cling' [35]. The dye-labeled PCR products were genotyped on a 3500 Genetic Analyzer (Life Technologies, Carlsbad, CA). Gene Marker 2.4 (Soft Genetics, State College, PA) was used to analyze the chromatographic trace files and generate the microsatellite allele table. The allele table generated from the four peach cultivars was converted to the format required by Power Marker [36] and imported into the program to calculate the allele number, heterozygosity value, polymorphism information content (PIC) value, and gene diversity value of each marker (ESM Table 2) and compare the differences between the 96 HPM and 96 HNM primers. A Student' st-test of "two-sample assuming unequal variances" was performed using the statistical add-ins in Excel Analysis Tool Pak (Microsoft, Seattle, WA) to determine whether all the four independent mean values between the HPM and HNM primers were significantly different at α=0.05, at which the t critical two-tail value is equal to 1.97 (ESM Table 2).
Functional analysis of HPM containing contigs
The sequences of contigs containing polymorphic microsatellites were submitted to BLAST2GO [37] for BLAST and gene ontology (GO) term annotation to predict the molecular function, biological process involved, and/or cellular component of the gene product.
Results
HPM among Prunus ESTs
A total of 319 contigs derived from 776 ESTs contained HPM (Table 2, ESM Table1), which were about 2.53% of 12,618 contigs assembled from 84,727 redundant ESTs. Of the 319 contigs, 262 possessed microsatellite polymorphisms between two representative EST haplotypes, and 57 contained microsatellite polymorphisms among more than two representative EST haplotypes (Table 2). Seven of the 57 contigs contained two or more polymorphic microsatellite motifs, which were flanked by different primer pairs for different amplicons and considered as different loci within the same contigs. Summarized from the view of species, 127 (39.8%) were found only within ESTs of peach, 108 (33.8%) were found in ESTs between peach and other species, and the remaining 84 were polymorphic in non-peach species (Table 2). However, there might be HPM from peach in the 84 non-peach HPM because redundant peach ESTs sharing the same polymorphic microsatellites with non-peach species were eliminated first.
![]()
Table 2: Species sources of ESTs containing polymorphic microsatellitesc
View Table 2
The 319 HPM were categorized in terms of their different motif types (Figure 2A). As expected, the p2, p3, and compound types were among top, accounting for 32.9%, 21.6%, and 17.6%, respectively. The p4, p5, and p6 types were minor, less than 5% each. Intriguingly, about 19.4% were the variant type, suggesting a substantial portion of other nucleotide variations such as transition, transversion, deletion, or insertion, in addition to usual repeat number differences, also occurred in these polymorphic microsatellite motifs. The differences between the repeat numbers in the HPM varied in the five basic motif types (Figure 2B), going up to seventeen in the p2 type, ranging from one to five in the p3 type, and remaining only one in the p4, p5, and p6 types.
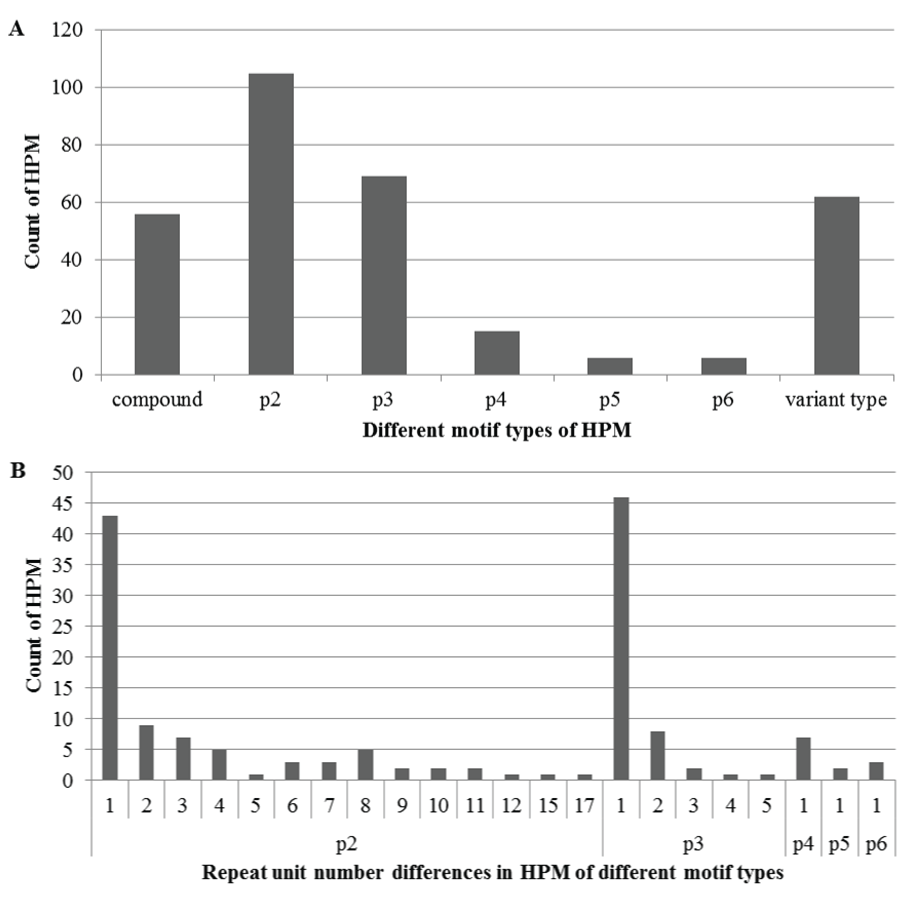
Figure 2: Different motif types of haplotype-based polymorphic microsatellites (HPM) (A) and repeat unit number differences in the HPM (B). In the x-axis of A, p2, p3, p4, p5, and p6 refers to motif types with a repeat unit length of two, three, four, five and six nucleotides, respectively; "compound" refers to the motif type with more than two of the five types adjacent in an interval length of 100bp; and "variant type" refers to different motif units derived from nucleotide complementarity or discrepancies .In the x-axis of B, each value is the difference of repeat unit numbers in each motif type. Each value in both y-axes refers a count of HPM in each type.
View Figure 2
Among all the 319 polymorphic contigs, Contig5674 contained the most polymorphic microsatellite motifs with at least 9 different numbers or variants of "CAT", the microsatellite unit in the representative ESTs (Figure 3A,B; ESM Table 1). In addition, there were two 6-nucleotide repeat units in the last 2 representative ESTs within the contig. According to the multiple alignments among representative ESTs (Figure 3A), the polymorphisms of this microsatellite motif apparently were due to deletions of individual nucleotides causing loss of some repeat units, rather than the deletion/addition of entire 3-nucleotid repeat units. Different sizes of amplicons were due to different numbers of repeat unit and/or variants of the microsatellite motif in the representative ESTs. The last amplicons covered the 6-nucleotide repeats in AM290100 and DW357109 (Figure 3B).
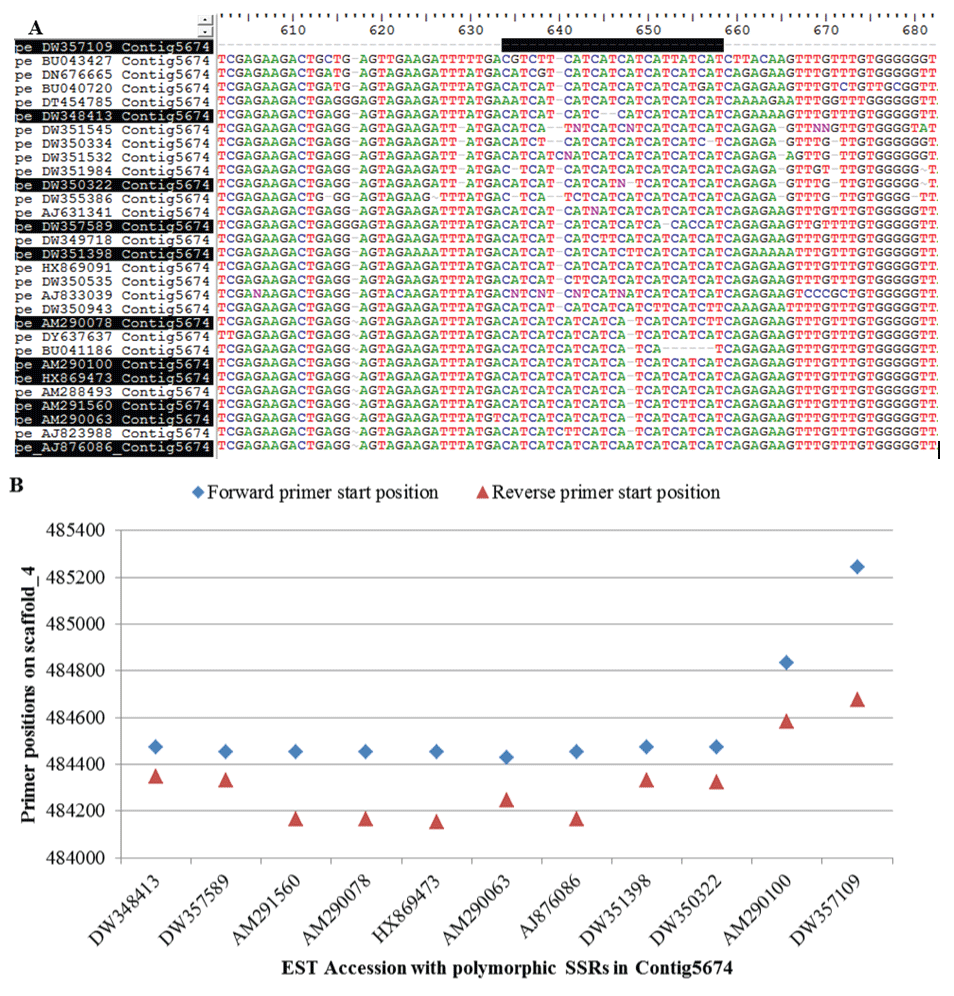
Figure 3: Alignment of representative polymorphic ESTs in Contig5674 (A) and their primer positions on peach scaffold_4 (B). The polymorphic "CAT" motifs start at 634 and end at 658 in the consensus sequence, which is marked by a black bar above the ESTs. These ESTs and their accession IDs highlighted in black in (A) are selected to show their forward and reverse primer start positions on peach scaffold_4 in (B). AM290100 and DW357109 in (B), not shown in (A), flank the other two 6-nucleotide microsatellites in the unigene.
View Figure 3
Microsatellite motifs and sequence alignment revealed micro satellite polymorphisms among the ESTs of the 319 contigs were primarily derived from different numbers of repeat unit of the same microsatellite motifs in the contigs (ESM Table 1). Different repeat variants and other nucleotide variations within some microsatellite motifs and/or amplicons were additional or standalone forms of polymorphisms, as demonstrated in Figure 3A. These variations increase the likelihood of microsatellite polymorphisms and also explain that the amplicon length differences may not always equal a multiple of the microsatellite motif unit length. The 319 primers were grouped into 7 categories by the ASD (Table 3) to indicate the calculated amplicon size ranges, possible amplification reliability, and other genomic features to help selection of primers of better performance.
![]()
Table 3: Categories of primers from polymorphic contigs and their assembled ESTsd
View Table 3
Genomic distribution of polymorphic microsatellites
The distribution of polymorphic microsatellites was generally scattered in the 8 main and several minor scaffolds of the peach reference genome (Figure 4). There were 69, 30, 39, 52, 33, 37, 31, and 29 polymorphic microsatellites in the 8 main scaffolds respectively, and a total of 8 in four minor scaffolds. On average, there was one polymorphic microsatellite every 678-kb genome region. It appeared fewer polymorphic microsatellites were distributed in certain regions of the scaffolds, one of which might belong to the centromere of the chromosomes, a region that generally contains highly repetitive DNAs and fewer genes.
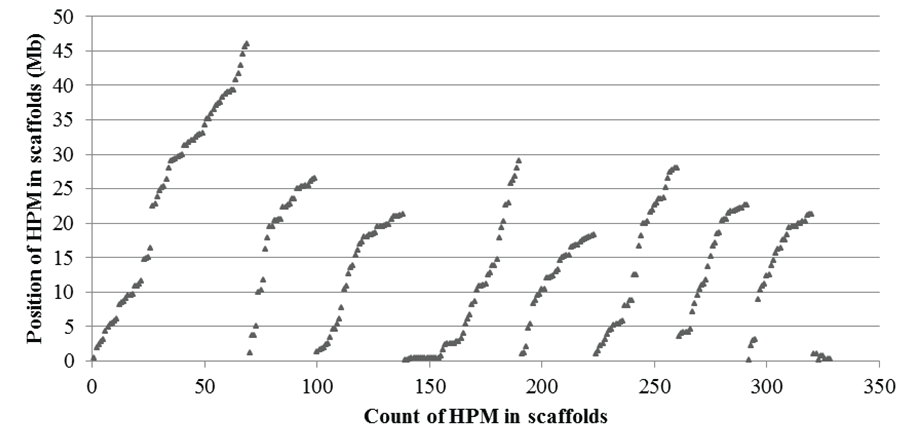
Figure 4: Distribution of haplotype-based polymorphic microsatellites (HPM) in 8 main (the first 8 groups) and several minor (the last group) scaffolds of the peach reference genome (v1.0). Among these HPM, only one EST for each polymorphic locus was used to display the distribution. The position of each HPM on a scaffold was represented by the start point of the forward primer sequence aligned onto the genome. There were 69, 30, 39, 52, 33, 37, 31, and 29 HPM in the 8 main scaffolds respectively, and a total of 8 in four minor scaffolds.
View Figure 4
Polymorphism rate comparison between primers of different categories and types
Only two primers, GW871526 (CX2H03) from the HPM and DW343006 (CX3E08) from the HNM, were failed in amplification and/or detection (ESM Table 2). The high success rate demonstrated that primers from the four categories with desired ASD and full primer alignment on the reference genome were highly reliable in genotyping. The number of alleles detected in the four peach genotypes ranged from 1 to 7 in both the HPM and HNM primers (Figure 5A,B). Differences in the average allele numbers and values of heterozygosity, polymorphism information content (PIC), and gene diversity were also observed among primers of the four different categories (Table 4) and between the HPM and HNM types (Table 5, Figure 6A-D, ESM Table 2). No statistical analysis was performed to compare the differences because the numbers of the primers in the four categories were so uneven, which resulted from random selection, along with a consideration of relatively even distribution on eight scaffolds of the peach reference genome. However, comparing the categories with more than ten primers, the primers in the "deletion" category in both HPM and HNM tended to yield the highest averages of allele number, heterozygosity value, and PIC value (Table 4), implying utilization of more primers in the category could increase the likelihood of polymorphism and heterozygosity. A comparison based on a larger, evener number of primers from these categories is needed for the validation.
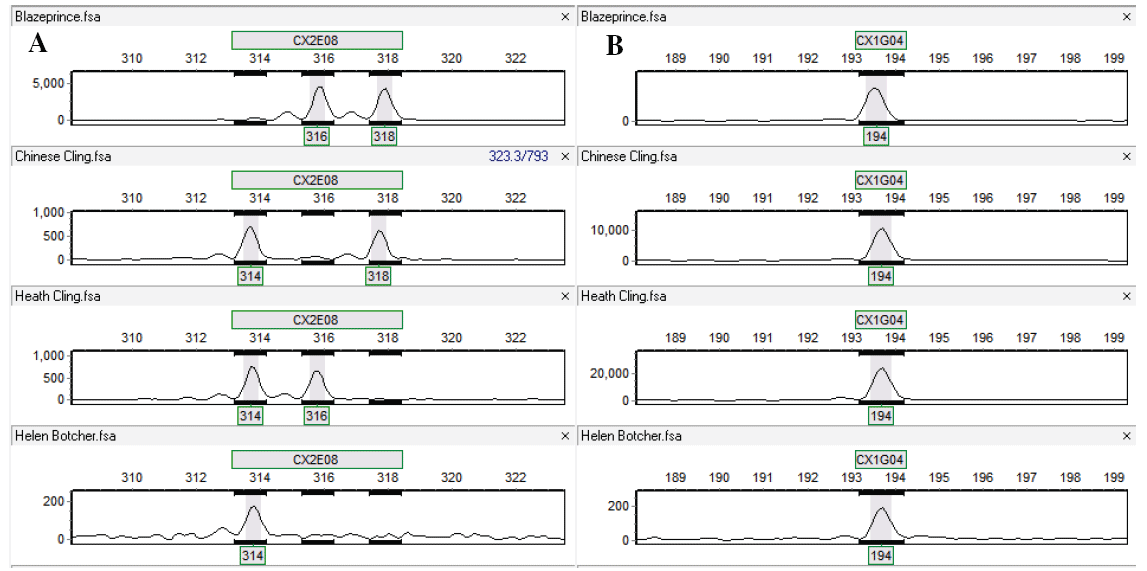
Figure 5: Typical microsatellite genotyping chromatographs with full polymorphisms (A) and no polymorphism (B) in the genotypes. The primers were CX2E08 and CX1G04. The four peach cultivars in turn were 'Blazeprince', 'Chinese Cling', 'Heath Cling', and 'Helen Borchers'. The relative size of amplicons was marked under each peak (allele). A single peak or two peaks in a genotype indicated the locus was homozygous or heterozygous, respectively.
View Figure 5
![]()
Table 4: Average allele numbers and values of heterozygosity, polymorphism information content (PIC), and gene diversity among primers of different categories
View Table 4
In general, the ranges of allele numbers and values of heterozygosity, PIC, and gene diversity for both HPM and HNM primers were similar or the same, but substantially more HPM primers yielded larger numbers and values, compared to the HNM primers (ESM Table 2). The four mean values calculated from all the HPM primers were marginally higher compared with those from the HNM primers among the 4 genotypes, but the differences were all statistically significant (P=0.003, 0.014, 0.002, and 0.002, respectively, ≤ α=0.05) (Table 5, ESM Table 2). In detail, there was a higher percentage of HPM markers with 4, 5, or 7 alleles detected, but a higher or equal percentage of HNM markers with the other numbers of alleles detected (Figure 6A). A higher percentage of HPM primers had heterozygosity values of 0.50, 0.67, and 1, but a lower or equal percentage of HPM primers had heterozygosity values of 0, 0.25, and 0.33, compared to HNM primers (Figure 6B). A higher percentage of HPM primers had PIC values of 0.7, 0.8 and 0.9, and was slightly lower at all other PIC values, compared to HNM primers (Figure 6C). A higher percentage of HPM primers had gene diversity values of 0.5, 0.8, and 0.9, but lower at all other gene diversity values observed (Figure 6D). The use of HPM primers could substantially increase the percentage of microsatellite markers with overall improvement of polymorphism and heterozygosity, which benefit these marker studies demanding allelic polymorphism and heterozygosity.
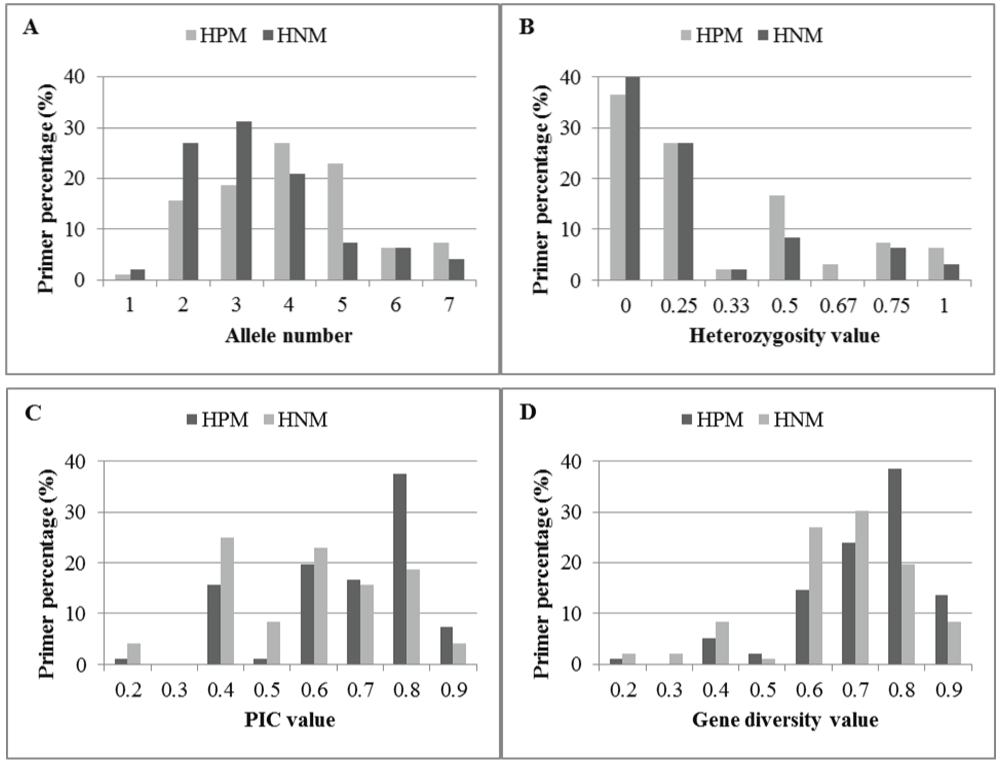
Figure 6: Haplotype-based polymorphic (HPM) and non-polymorphic (HNM) primer percentages in allele numbers (A) heterozygosity values (B) PIC values (C) and gene diversity values (D) obtained from 4 peach cultivars. Each x-axis value in C and D represents the upper bound of a range, for example, "0.2" is "<=0.2", "0.3" is >0.2 &<=0.3, and so on.
View Figure 6
![]()
Table 5: t-test for the mean allele numbers and values of heterozygosity, PIC, and gene diversity between the HPM and HNM primersg
View Table 5
Annotation of polymorphic microsatellites contained unigenes
Of the 319 polymorphic contigs, only 234 were annotated with GO molecular function terms by BLAST2GO. Ninety-nine unigenes (42.3%) were categorized to have binding function and 84 (35.9%) to have catalytic activity (Figure 7), implying that these polymorphic alleles might have evolved primarily to play regulatory (binding) roles in gene expression, or to catalyze enzymatic reactions in various biosynthesis pathways. Unlike the predominant categorization into the binding and catalytic activities based on the GO molecular function terms, unigenes were distributed relatively evenly in the annotated biological process or cellular component GO terms (data not shown). Contig5674, the unigenes containing the most polymorphic microsatellite motifs, was not annotated by BLAST2GO. However, a BLASTN search of Contig5674 to the National Center for Biotechnology Information (NCBI) non-redundant nucleotide database revealed that it shared very high nucleotide identities with a putative allergen protein gene in several Prunus species and other plant genera. An allergen gene usually has more frequent nucleotide changes to maintain the allergen function over highly variable antibodies, which could explain to some extent the exceptionally high nucleotide variation rate in the motif.
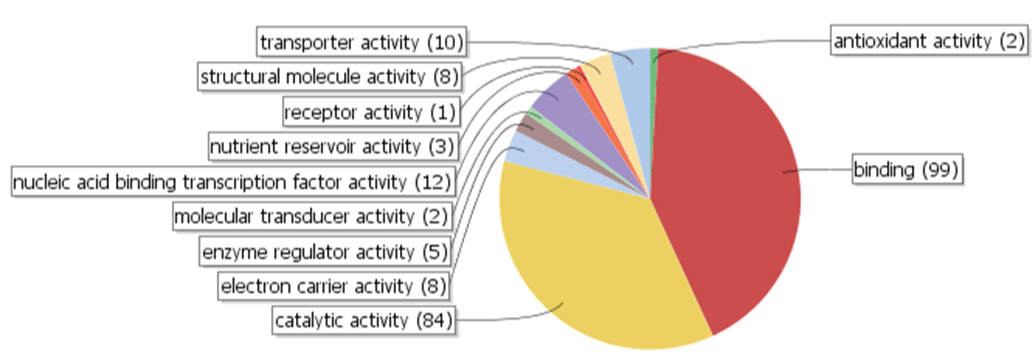
Figure 7: Polymorphic unigenes categorized by BLAST2GO, according to molecular function gene ontology (GO) terms used by the program. Binding and catalytic activity are the two principal categories in the 234 annotated unigenes, accounting for 99 (42.3%) and 84 (35.9%) of the unigenes, respectively.
View Figure 7
Discussion
Variations in HPM motifs and amplicons
Although microsatellite polymorphisms are based on amplicons of different lengths generally derived from varying repeat numbers of microsatellite units [5,11], this study revealed additional nucleotide variations occurred in some polymorphic motifs and/or amplicon regions (Figure 2A,3A), including SNPs. These additional nucleotide variations in an HPM motif and/or amplicon region not only increase the likelihood of the microsatellite polymorphism, but also partly explain that some length difference among detected polymorphic alleles may not always equal a multiple of the unit length of the microsatellite motif. These SNPs within microsatellite motifs and amplicons could independently be mined for SNP development [6,7]. On the other hand, although microsatellites and their flanking primers are relatively easy to mine out from vast expressed and genomic sequences [5], utilization of randomly selected microsatellite primers has led to a high rate of failure largely due to unknown sizes of introns in amplicons and/or primers from erroneous or poor-quality regions of source sequences [9]. Therefore, the seven categorizations (Table 3) are helpful for selection of more reliable primers with better chromosomal distribution (Figure 4). Predictably, the primers in the "deletion", "same size", "insertion", and "intron (GAS<=500)" are much more reliable in amplification and detection than those in the "NHF", "error", and "intron (GAS>500)" categories.
Allelic functions and polymorphisms
Interestingly, of the 234 annotated unigenes, 99 (~42.3%) were categorized to have binding function and 84 (~35.9%) to have catalytic activity (Figure 7). The polymorphic alleles or genes found associated with the microsatellites in this study apparently play various regulatory (binding) and enzymatic (catalysis) roles, which may contribute substantially to the diversification of Prunus species and cultivars [34]. These HPM markers, representing partly some of the most variable alleles (genes) or gene families, could be more valuable in future studies of Prunus evolution and domestication. Primer sequences failed to align onto the peach reference genome among the 46 NHF contigs might be additional evidence that high nucleotide variability in these highly polymorphic alleles, unless some sequencing and/or assembly errors existed in these EST sequences or to-be-aligned genomic regions. Of the 319unigenes containing HPM, 127 (~39.8%) were from ESTs only in peach, 108 (~33.8%) were from ESTs in peach and other species, and the remaining 84 were from ESTs from non-peach species. The distribution of polymorphisms among these species might not reflect the true polymorphism status among them due to unbalanced numbers of ESTs used in the study. On the other hand, the polymorphism rates within peach could be higher because redundant ESTs from peach were eliminated first and the priority to keep an EST to represent each unique polymorphic microsatellite motif was given to non-peach species. Increasing EST number and genome coverage for Prunus species of greatest interest would yield an improved estimation on their polymorphism status across the species [26]. According to the comparison of the HPM and HNM primers in this study, a significantly higher mean allele number and values of heterozygosity, PIC, and gene diversity were detected in four relatively close peach genotypes, suggesting a substantially higher rate of polymorphism and heterozygosity among the HPM primers, compared to the HNM primers (Table 5, Figure 6A-D). It obviously was due to more HPM primers than HNM primers yielding the higher ranges of the four values. The improvement of allele polymorphism and heterozygosity certainly would be expected among relatively distant Prunus species, as the transportability of previous primers from some of these species had been proved [19,20]. Further investigation into these species is needed (Supplementary Table 1).
Acknowledgements
The authors thank Minling Zhang, Bryan Blackburn, and Luke Quick for their technical assistance. The research is partially supported by the USDA National Program of Plant Genetic Resources, Genomics and Genetic Improvement (Project number: 6606-21000-004-00D).
This article reports the results of research only. Mention of a trademark or proprietary product is solely for the purpose of providing specific information and does not constitute a guarantee or warranty of the product by the U.S. Department of Agriculture and does not imply its approval to the exclusion of other products that may also be suitable.
References
-
Aranzana MJ, Carbo J, Arus P (2003) Microsatellite variability in peach [Prunus persica (L.) Batsch]: cultivar identification, marker mutation, pedigree inferences and population structure. Theor Appl Genet 106: 1341-1352.
-
Ahmad R, Potter D, Southwick SM (2004) Genotyping of peach and nectarine cultivars with SSR and SRAP molecular markers. J Am Soc Hort Sci 129: 204-210.
-
Chen C, Bowman KD, Choi YA, Dang PM, Nageswara Rao D, et al. (2008) EST-SSR genetic maps for Citrus sinensis and Poncirus trifoliata. Tree Genet Genomes 4: 1-10.
-
Jones N, Ougham H, Thomas H, Pasakinskiene I (2009) Markers and mapping revisited: finding your gene. New Phytol 183: 935-966.
-
Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106: 411-422.
-
Verde I, Bassil N, Scalabrin S, Gilmore B, Lawley CT, et al. (2012) Development and evaluation of a 9K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm. PLoS One 7: e35668.
-
Tang J, Vosman B, Voorrips RE, van der Linden CG, Leunissen JA (2006) Quality SNP: a pipeline for detecting single nucleotide polymorphisms and insertions/deletions in EST data from diploid and polyploid species. BMC Bioinformatics 7: 438.
-
Ahmad R, Parfitt DE, Fass J, Ogundiwin E, Dhingra A, et al. (2011) Whole genome sequencing of peach (Prunus persica L.) for SNP identification and selection. BMC Genomics 12: 569.
-
Chen C, Bock CH, Beckman TG (2014) Sequence analysis reveals genomic factors affecting EST-SSR primer performance and polymorphism. Mol Genet Genomics 289: 1147-1156.
-
Warnich L, Groenewald I, Laubscher L, Retief AE (1991) Improvement of polymorphic information content of DNA markers by exploring microsatellite sequences. Am J Hum Genet 49: 372-372.
-
Kong Q, Zhang G, Chen W, Zhang Z, Zou X (2012) Identification and development of polymorphic EST-SSR markers by sequence alignment in pepper, Capsicum annuum (Solanaceae). Am J Bot 99: e59-61.
-
Kayesh E, Zhang YY, Liu GS, Bilkish N, Sun X, et al. (2013) Development of highly polymorphic EST-SSR markers and segregation in F? hybrid population of Vitis vinifera L. Genet Mol Res 12: 3871-3878.
-
Mohanty P, Sahoo L, Parida K, Das P (2013) Development of polymorphic EST-SSR markers in Macrobrachium rosenbergii by data mining. Conserv Genet Resour 5: 133-136.
-
Bizzaro JW, Marx KA (2003) Poly: a quantitative analysis tool for simple sequence repeat (SSR) tracts in DNA. BMC Bioinformatics 4: 22.
-
Cipriani G, Lot G, Huang WG, Marrazzo MT, Peterlunger E, et al. (1999) AC/GT and AG/CT microsatellite repeats in peach [Prunus persica (L) Batsch]: isolation, characterisation and cross-species amplification in Prunus. Theor Appl Genet 99: 65-72.
-
Sosinski B, Gannavarapu M, Hager LD, Beck LE, King GJ, et al. (2000) Characterization of microsatellite markers in peach [Prunus persica (L.) Batsch]. Theor Appl Genet 101: 421-428.
-
Testolin R, Marrazzo T, Cipriani G, Quarta R, Verde I, et al. (2000) Microsatellite DNA in peach (Prunus persica L. Batsch) and its use in fingerprinting and testing the genetic origin of cultivars. Genome 43: 512-520.
-
Dirlewanger E, Cosson P, Tavaud M, Aranzana J, Poizat C, et al. (2002) Development of microsatellite markers in peach [ Prunus persica (L.) Batsch] and their use in genetic diversity analysis in peach and sweet cherry (Prunus avium L.). Theor Appl Genet 105: 127-138.
-
Mnejja M, Garcia-Mas J, Howad W, Arus P (2005) Development and transportability across Prunus species of 42 polymorphic almond microsatellites. Mol Ecol Notes 5: 531-535.
-
Vendramin E, Dettori MT, Giovinazzi J, Micali S, Quarta R, et al. (2007) A set of EST-SSRs isolated from peach fruit transcriptome and their transportability across Prunus species. Mol Ecol Notes 7: 307-310.
-
Wang Y, Georgi LL, Zhebentyayeva TN, Reighard GL, Scorza R, et al. (2002) High-throughput targeted SSR marker development in peach (Prunus persica). Genome 45: 319-328.
-
Dirlewanger E, Cosson P, Howad W, Capdeville G, Bosselut N, et al. (2004) Microsatellite genetic linkage maps of myrobalan plum and an almond-peach hybrid--location of root-knot nematode resistance genes. Theor Appl Genet 109: 827-838.
-
Howad W, Yamamoto T, Dirlewanger E, Testolin R, Cosson P, et al. (2005) Mapping with a few plants: using selective mapping for microsatellite saturation of the Prunus reference map. Genetics 171: 1305-1309.
-
Verde I, Lauria M, Dettori MT, Vendramin E, Balconi C, et al. (2005) Microsatellite and AFLP markers in the Prunus persica [L. (Batsch)]xP. ferganensis BC(1)linkage map: saturation and coverage improvement. Theor Appl Genet 111: 1013-1021.
-
Liu X, Reighard G, Swire-Clark GA, Bridges WC, Abbott AG, et al. (2009) Identification of the chromosomal genomic regions associated with peach tree short life syndrome using microsatellite/SSR markers. HortSci 44:1175-1176.
-
Jung S, Abbott A, Jesudurai C, Tomkins J, Main D (2005) Frequency, type, distribution and annotation of simple sequence repeats in Rosaceae ESTs. Funct Integr Genomics 5: 136-143.
-
Aranzana MJ, Illa E, Howad W, Arus P (2012) A first insight into peach [Prunus persica (L.) Batsch] SNP variability. Tree Genet Genomes 8: 1359-1369.
-
Sánchez G, Martínez J, Romeu J, García J, Monforte AJ, et al. (2014) The peach volatilome modularity is reflected at the genetic and environmental response levels in a QTL mapping population. BMC Plant Biol 14: 137.
-
Sonah H, Bastien M, Iquira E, Tardivel A, Légaré G, et al. (2013) An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS One 8: e54603.
-
Chen X, Sullivan PF (2003) Single nucleotide polymorphism genotyping: biochemistry, protocol, cost and throughput. Pharmacogenomics J 3: 77-96.
-
Thalamuthu A, Mukhopadhyay I, Ray A, Weeks DE (2005) A comparison between microsatellite and single-nucleotide polymorphism markers with respect to two measures of information content. BMC Genet 6 Suppl 1: S27.
-
Morgante M, Hanafey M, Powell W (2002) Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat Genet 30: 194-200.
-
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132: 365-386.
-
Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, et al. (2013) The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet 45: 487-494.
-
Okie WR (1998) Handbook of Peach and Nectarine Varieties: Performance in the Southeastern United States and Index of Names. The National Technical Information Service, Springfield, VA
-
Liu K, Muse SV (2005) Power Marker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21: 2128-2129.
-
Conesa A, Götz S (2008) Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008: 619832.
-
Gordon D, Desmarais C, Green P (2001) Automated finishing with autofinish. Genome Res 11: 614-625.
-
Huang X, Madan A (1999) CAP3: A DNA sequence assembly program. Genome Res 9: 868-877.
-
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389-3402.
