

International Journal of Cancer and Clinical Research
Developing Pathway Collection for Personalized Anti-cancer Therapy
Luminita Castillos1 and Anton Yuryev2*
1Personalized Hematology-Oncology of Wake Forest, USA
2Biology Elsevier Life Sciences Solutions, Rockville, USA
*Corresponding author:
Anton Yuryev, Professional Services Director, Biology Elsevier Life Sciences Solutions, 5635 Fishers Lane, Suite 510, Rockville, MD 20852, USA, Tel: +1-240-221-4507, Fax: 240-221-2540, E-mail: A.Yuryev@elsevier.com
Int J Cancer Clin Res, IJCCR-3-043, (Volume 3, Issue 1), Research Article; ISSN: 2378-3419
Received: November 17, 2015 | Accepted: January 30, 2016 | Published: February 01, 2016
Citation: Castillos L, Yuryev A (2016) Developing Pathway Collection for Personalized Anti-cancer Therapy. Int J Cancer Clin Res 3:043. 10.23937/2378-3419/3/1/1043
Copyright: © 2016 Castillos L, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
We describe methodology for developing personalized anti-cancer drug therapy using pathway analysis. We successfully applied this methodology to treat several cancer patients that were terminally diagnosed by standard of care criteria at the hospital. Our approach consists of profiling patient tumor using gene expression microarray and calculating pathways responsible for the differential expression between tumor and normal control tissue. Pathways are selected for treatment targeting based on their enrichment with major expression regulators identified by sub-network enrichment analysis (SNEA) in Pathway Studio. We then select FDA approved drugs inhibiting activated pathways and prescribe them to the patient. To facilitate interpretation of patient data we built collection of cancer pathways based on ten cancer hallmarks described in the literature. This collection explains function of more than half of expression regulators identified in patient’s tumors by SNEA. This paper focuses on description of pathways built for interpretation of expression profiles of cancer patients.
Introduction
More than 400 FDA approved drugs are currently on the market and were in clinical trials for treatment of common types of cancer in the last five years. Most of these drugs have known mechanism of action and directly target more than 800 proteins in human genome. While not every clinical trial is successful the number of treatment options for cancer is already large and only expected to grow in the future. Thus, the major challenge for modern oncologists is selecting the most effective anti-cancer treatment for a patient from the vast number of approved anti-cancer drugs on the market. The proposed solution for this problem is called personalized medicine - development of approaches for selecting the best treatment for a patient based on drug mechanism of action that most optimally matches the molecular mechanism driving tumor growth.
Gene expression microarray technology is the oldest and most robust method for large scale molecular profiling of cancer patients [1,2]. While detection technology was improving in last two decades analytical methods were developed for calculating differentially expressed (DE) genes and transcriptional signatures from DE genes from the limited number of patients. These computational approaches yielded a lot of insights into cancer biology [3] but also revealed "curse of dimensionality" of the large scale molecular profiling data [4]. "Curse of dimensionality" is the contradiction between mathematical requirement for optimal gene signatures to contain no more than 20-30 genes [5] and biological reality observing10-100 times more differentially expressed genes in cancer tumors. Such short optimal signature are the consequence of the small number of patient samples available in a training set for signature calculation compared to the number of correlated DE genes [6,7]. The shortage of cancer samples for large signature calculation is so significant that even 10-fold increase in the number of available samples will not yield substantial improvement in predictability of transcriptional signatures.
Biological considerations can provide solution to the "curse of dimensionality" of microarray data. Indeed, observed transcriptional profile in a patient is due to the activity of transcription factors and micro RNAs. The number of these direct transcriptional regulators is much smaller than the number of genes on the microarray and in human genome. Thus, the transformation of transcriptional profile into activity of few upstream expression regulators should provide significant reduction in the data space dimensionality which in turn should help calculating more powerful signatures [8]. Two similar algorithms were developed to calculate the activity of upstream expression regulators from microarray data using prior knowledge about expression regulatory events reported in the literature: sub-network enrichment analysis (SNEA) [9] and reverse causal reasoning (RCR) [10]. We used SNEA algorithm implemented in Pathway Studio software from Elsevier. It relies on the knowledge base of expression regulation events automatically extracted from biomedical research literature by natural processing technology. Pathway Studio knowledge base has the biggest number of regulatory events and therefore provides the most comprehensive and up-to-date snapshot of transcriptional activity in cancer samples. SNEA uses non-parametric Mann-Whitney enrichment test to evaluate transcriptional activity of upstream regulators which was shown to provide superior results for microarray data analysis over overlap hyper geometric test implemented in RCR [11,12].
The activity of upstream expression regulators in turn depends on activity of pathways altered in the tumor. Therefore projecting the activity of upstream expression regulators identified by SNEA onto collection of relatively small number of biological pathways relevant for cancer progression should allow us to identify cancer mechanism in an individual patient reducing the complexity in interpretation of large number of differentially expressed genes in a tumor. A set of ten high-level biological processes responsible for cancer progression is known as hallmarks was suggested [13]. Each cancer hallmark can be facilitated by several alternative mechanisms described in the literature but in one given patient only one of these alternatives is realized by the tumor at one time. Because the same biological mechanism may still involve different proteins in different tissues several hundred pathways have to be built in order to create a comprehensive collection of cancer hallmarks mechanisms in different tissues. Yet, only a handful of these pathways will be activated in a single patient.
In this article we describe workflow for personalized medicine that uses gene expression profile of patient tumor to identify major expression regulators which transcriptional activity is significantly altered in the tumor according to SNEA. Using most significant expression regulators identified from five transcriptional tumor profiles of three cancer patients we have constructed the library of cancer hallmark pathways. All pathways are based on translational data from scientific literature that studied cancer cell lines or cancer in animal models. These pathways can explain activity of about 60% expression regulators in our patients. We assert that pathways enriched with major SNEA regulators have altered activity in the tumor and therefore should be used to select drugs for personalized treatment. We validated our approach by selecting drugs inhibiting the activity of identified pathways. Even though sometimes it was not possible to find drug that could directly inhibit expression regulators found by SNEA drugs selected based on upstream pathway information prolonged patient survival beyond Overall Survival estimates based on standard of care treatment. This article focuses on describing pathways identified by our approach and used for treatment selection.
Materials and Methods
Patients
First patient (liver cancer)
• 66 year old Caucasian female diagnosed with moderate to poorly differentiated hepatocellular carcinoma with associated necrosis
• Pet/Ct scan shows 9.0 × 7.2 × 5.7 cm right hepatic lobe mass
• Resection of hepatocellular carcinoma involving the ascending colon in the right lateral abdominal wall, segments 5 and 6 from the liver and 11 benign lymph nodes
• Core biopsies of liver tumor as well as some of the normal liver parenchymal cells were sent for gene expression profile analysis.
Second patient (breast cancer)
• 66 year old Caucasian female diagnosed in 2011 in Florida with stage I breast cancer, miss-labeled as ER+/PR+, and treated with Docetaxel/Cyclophosphamide (4 cycles) followed by hormonal therapy
• Cancer recurrence in 2013 and diagnosed with stage IV breast cancer with metastasis in the right lung and her brain (diagnose made in our practice for the first time after moving to North Carolina)
• Re-diagnosed (initial tumor block from Florida) as ER-/PR- and treated with radiation for the brain metastasis and 2 cycles of Adriamycin/Cyclophosphamide (dose dense standard of care therapy, based on ASCO and NCCN guidelines ) for the breast cancer lung mets
• Brain metastasis responded to the radiation treatment but the lung metastasis did not respond to AC chemotherapy and core biopsies were performed from the lung met
• Treatment was switched to Gemcitabine (standard of care) until the gene expression profiling data was processed
Third patient (colon cancer)
• 74 year old Caucasian male diagnosed in 2009 with stage IV colon cancer
• Removal of sigmoid colon
• Radiofrequency ablation for two liver lesions
• Treated for surgical site infection
• Refused to have chemotherapy (adjuvant therapy) initially after the surgery
• Cancer recurrence in 2013 with multiple mets in the liver and lung; core biopsies were performed from the liver met for gene expression profiling
• In January 2014 started standard of care modified FOLFOX6 regimen every 2 weeks with 5-FU CADD pump
• FOLFOX6 = 5-FU + Oxaliplatin + Leucovorin
Gene expression profile
Human U133 Plus 2.0 array were used to process the patients RNA samples. Also, the following instruments were used: Affymetrix Gene Titan instrument for processing the microarrays, GeneChip Hybridization oven 640, two Fluidics Station 450s, and Affymetrix Gene Chip Scanner 3000.
Calculation of differentially expressed genes in patient tumor biopsies
Typically, we were able to measure only one microarray profile for each patient tumor biopsy. Whenever possible we tried to collect syngenic samples to calculate differentially expressed genes. If syngenic sample was not available we used control samples from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/). For this, we downloaded from GEO all possible profiles of healthy human tissues that correspond to patient tumor tissue and measuring expression profile on the same microarray chip (HG-U133 plus 2.0 from Affymetrix). The same chip requirement allowed RMA normalization of CEL files for our patient samples together with samples downloaded for GEO.
For patient with colon cancer with metastasis in lung we used syngenic control from other healthy lung. For the patient with liver cancer we used syngenic control from healthy part of the liver. For patient with breast cancer metastasis in lung we used six normal breast tissue samples from GSE3744; for patient with colon cancer metastasis in liver we used 17 control samples from GSE32323; for patient with lung cancer we used 14 normal lung samples from GSE30219. Differentially expressed genes in all cases were calculated in Pathway Studio using unpaired t-test. P-value of differential expression for each probe on the array was possible to calculate only if patient data was normalized on multiple normal control samples from GEO. For these cases only probes with p-value less than 0.05 were used as input for calculation of SNEA regulators. For genes measured by several probes on HG-U133 plus 2.0 chip Pathway Studio selects the probe with best p-value for SNEA.
Pathway Studio: SNEA and cancer pathway reconstruction
We used Pathway Studio version 9 with knowledge base containing data extracted by Elsevier natural language processing (NLP) technology from all Pubmed abstracts and from more than 2,000,000 full-text articles published in about 1,200 biomedical journals [14]. Elsevier NLP extracts various types of biological interactions for Pathway Studio database. In order to identify upstream expression regulators by SNEA we used Expression and Promoter Binding regulatory interactions. These relation types are included in the option "Expression targets" in Pathway Studio menu for SNEA. Most regulators identified by this SNEA option are transcription factors, receptors, secreted hormones and extracellular matrix proteins. Typically, we were able to map about 30% of all SNEA regulators identified from patient expression profile on the signaling and cell process pathways that already existed in Pathway Studio, e.g. pathways for cell cycle, apoptosis regulation, DNA repair and chromatin remodeling. For purpose of building additional cancer pathways we connected remaining regulators that have not been mapped on existing pathways, with physical interactions in Pathway Studio database (i.e., Binding, Direct Regulation and Protein Modification) in order to find regulators involved in common pathway. Functionally-related regulators appeared as clusters in the physical interaction network. We then performed literature search to find articles reporting on the role of proteins in cancer in each network cluster. We preferred to use one or several review articles found by the literature search to reconstruct new cancer pathways in Pathway Studio (Table 1). If review articles were not available we used an original research articles that reported on the role of patient expression regulators in cancer.
![]()
Table 1: List of cancer hallmark pathways with supporting literature. PMID – Pubmed ID of the article used for pathway reconstruction. Cancer hallmark processes are borrowed from [13].
View Table 1
Pathway studio: drug selection
We used relations depicting drug effects extracted by Elsevier NLP ChemEffect cartridge [15] in order to find drugs for patient treatment. Our drug selection was done by following progressive steps. First, we attempted to find FDA-approved anti-cancer drugs inhibiting activated expression regulators that have been identified by SNEA. For this, we looked for drugs linked to SNEA regulators by Direct Regulation with Effect negative or Binding and then for drugs linked to SNEA regulators by Regulation with Effect negative. This approach proved to find only limited number of FDA approved drugs because many SNEA regulators were not druggable. In case the drugs inhibiting SNEA regulators were not available we tried to find drugs inhibiting activity of the cell process activated in a patient according to pathway analysis. The cell process regulated by one of the cancer pathways was usually also significant in SNEA with option using Cell Process as seed entity. This SNEA option finds Cell Process entities regulated by DE genes. Because DE genes regulating the cell process are downstream of the pathway regulating this cell process we observed correlation between results of two SNEA options: "Expression targets" and "Proteins regulating cell process". The drugs were selected by finding appropriate Cell Process entity (e.g. "Epithelial-to-mesenchymal transition" or "Cell invasion") in Pathway Studio and expanding it upstream towards Small Molecules with Regulation Effect negative. As additional selection criteria we preferred the drugs with demonstrated efficacy against the type of cancer developed in the analyzed patient. We could check known drug efficacy in Pathway Studio by looking up the relations between the drug and the right type of cancer with Regulation relation with Effect negative.
Statement of ethics
All but one patient were treated with FDA approved drugs prescribed by physician. Liver cancer patient was placed on a clinical trial of Sorafenib + Voronistat combination at the Massey Cancer Center in Richmond, Virginia after approval by its ethics committee.
Results
Pathway collection for analysis of cancer patients
We have constructed 49 pathways containing 2,779 proteins (Table 1). Pathways represent different mechanisms for ten cancer hallmark processes [13]. Pathways for cell cycle, apoptosis, DNA repair, chromatin remodeling, and EGFR signaling were copied from existing Pathway Studio pathway collection with minor modifications that added patient SNEA regulators involved in the same process. New pathways were built to depict the roles of found SNEA regulators in cancer as described in Materials & Methods section. The list of cancer pathways with supporting articles used for pathway construction is available in table 1. Several representative pathways are shown on figure 1. Our pathways explain function of 378 out of 600 top 100 SNEA regulators in every patient (63%). About 20% of remaining unexplained regulators were proteins involved in immune response. These proteins are likely to contribute to tumor-induced inflammation. We did not consider these proteins as therapeutic targets for our patients and therefore we do not describe them in this paper.
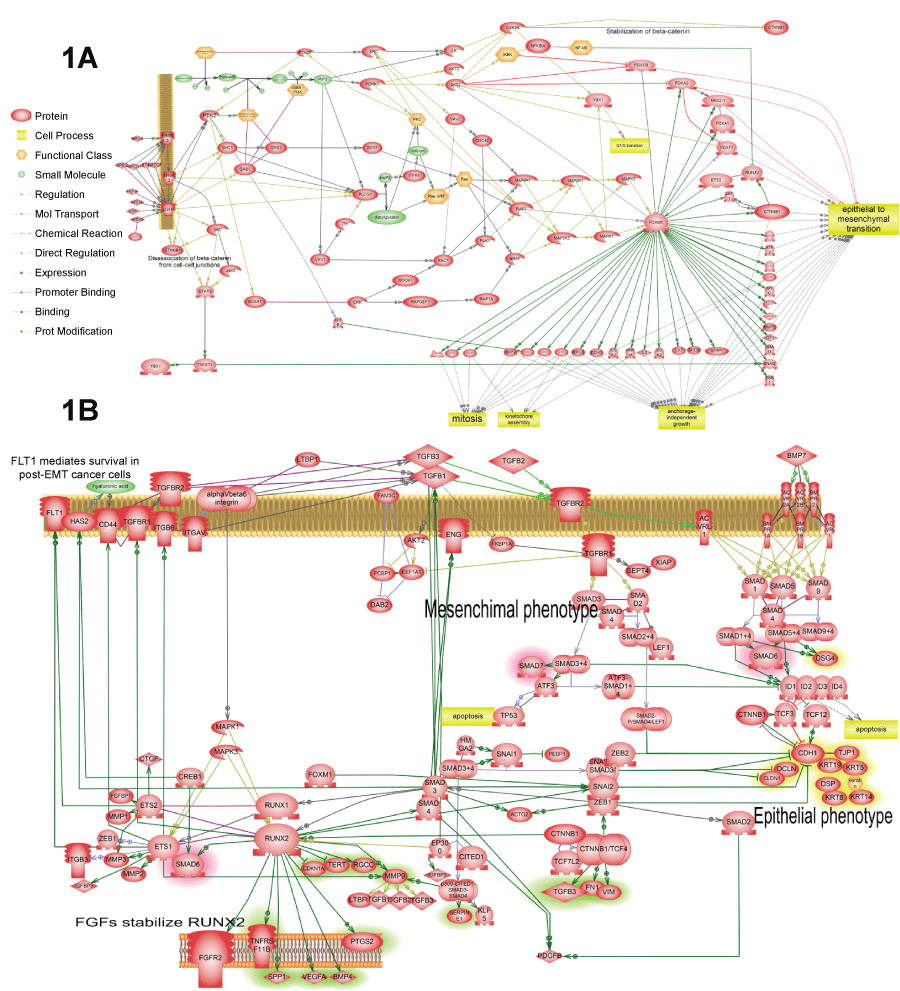
.
Figure 1: Four representative pathways that were found activated in several cancer patients according to our analysis. The complete list of such pathways is available in tables 2 and 3. All pathways were enriched with major expression regulators identified by SNEA with p-value smaller than 0.05 according to Fisher exact test.
A) Shows FoxM1 activation by EGFR. Pathway shows how FoxM1 protein is activated by EGFR and how FoxM1 activates cell proliferation and epithelial-to-mesenchymal transition; B) Shows TGF-beta autocrine loop that establishesmesenchymal state in the tumor cells. This pathway was found activated in all three patients (Table 3). The primary positive feedback loop represents almost canonical knowledge. It consists of TGF-beta expression activation by SMAD3/4 transcription factors, which are in turn induced by TGF-beta receptor signaling. Secondary positive feedback loop is novel finding based on the analysis of available literature about TGF-beta activation. It includes induction of RUNX1/2 by SMAD3/4 which then induces expression of TGF-beta receptors and integrins that increase binding of TGF-beta ligand to cell surface. Additionally, expression growth hormone receptors FGFR2 and FLT1 are induced to sustain cells in proliferative state that tis necessary for EMT. RUNX aldo induce transcription of angiogenic factor VEGF and extracellular matrix adhesion molecule CD44 necessary for cell migration;
View Figure 1A&B
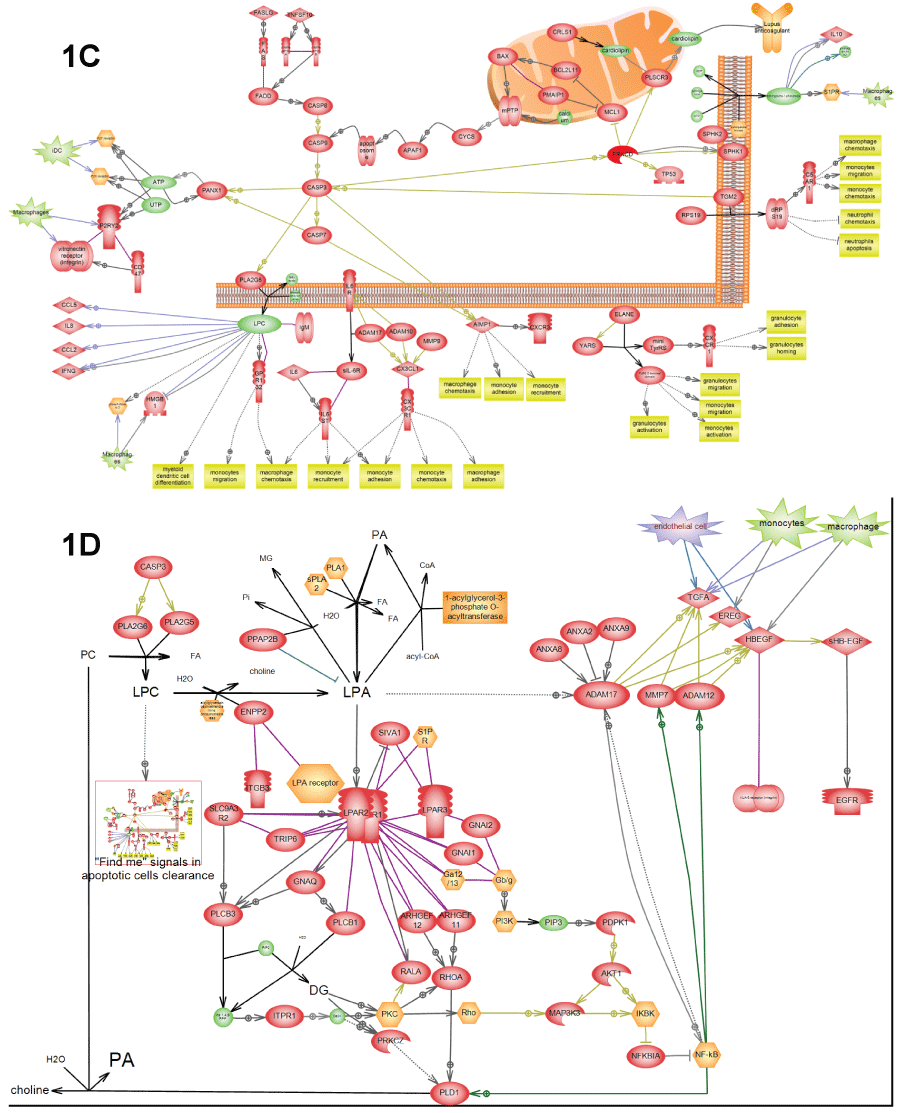
.
Figure 1: C) Depicts "Find me" signals" in apoptotic clearance – certain molecules in apoptotic debris that serve as signals for macrophage activation. This pathway was found activated in all three patients (Table 3); D) Depicts EGFR activation by apoptotic clearance. This pathway resembles the wound healing pathway and was found activated in all three patients (Table 3). "Find-me" signals from apoptotic debris (LPC) activate macrophages growth factors to secrete growth factors. LPC is also converted into LPA by secreted lysophospholipase D. LPA activates LPA receptor on the surface of epithelial cells that in-turn activate NF-kappa-B transcription factor in order to express and secrete matrix metalloproteases (e.g. ADAM17). These matrix metalloproteases convert growth factors secreted by macrophages into activated form and induce cell proliferation. Depiction of cells from adaptive immune response secreting EGFR ligands is based on the relatively new report [21] from 2013.
View Figure 1C&D
Identification of pathways with altered activity in cancer patients
Top 100 SNEA regulators with p-value less than 0.05 were determined in every patient tumor sample and compared with our cancer pathway collection to find pathways enriched with SNEA regulators. Statistical significance of enrichment was determined by Fisher’s exact test implanted in Pathway Studio menu option "Find similar pathways". Because many signaling pathways share common intracellular signaling components such as MAPK kinases and activation of stress or inflammatory response transcription factors (e.g. AP1, STATs, CREBs, NF-κB) we have used additional criteria to select activated pathways: the ligand or corresponding receptor initiating the pathway must be on the list of major regulators calculated by SNEA. The results for each patient are shown in table 2 and figure 2. We found that patient’s tumor profiles were more similar on the level of activated pathways then on the level of individual SNEA regulators. Table 3 lists 26 pathways that were found activated in all three patients. Patient differences on the level of SNEA regulators were most likely due to tissue specificity of gene expression since two patients with lung cancer had 72% of SNEA regulators in common while only 10% of regulators were common in patients with cancers from different tissues.
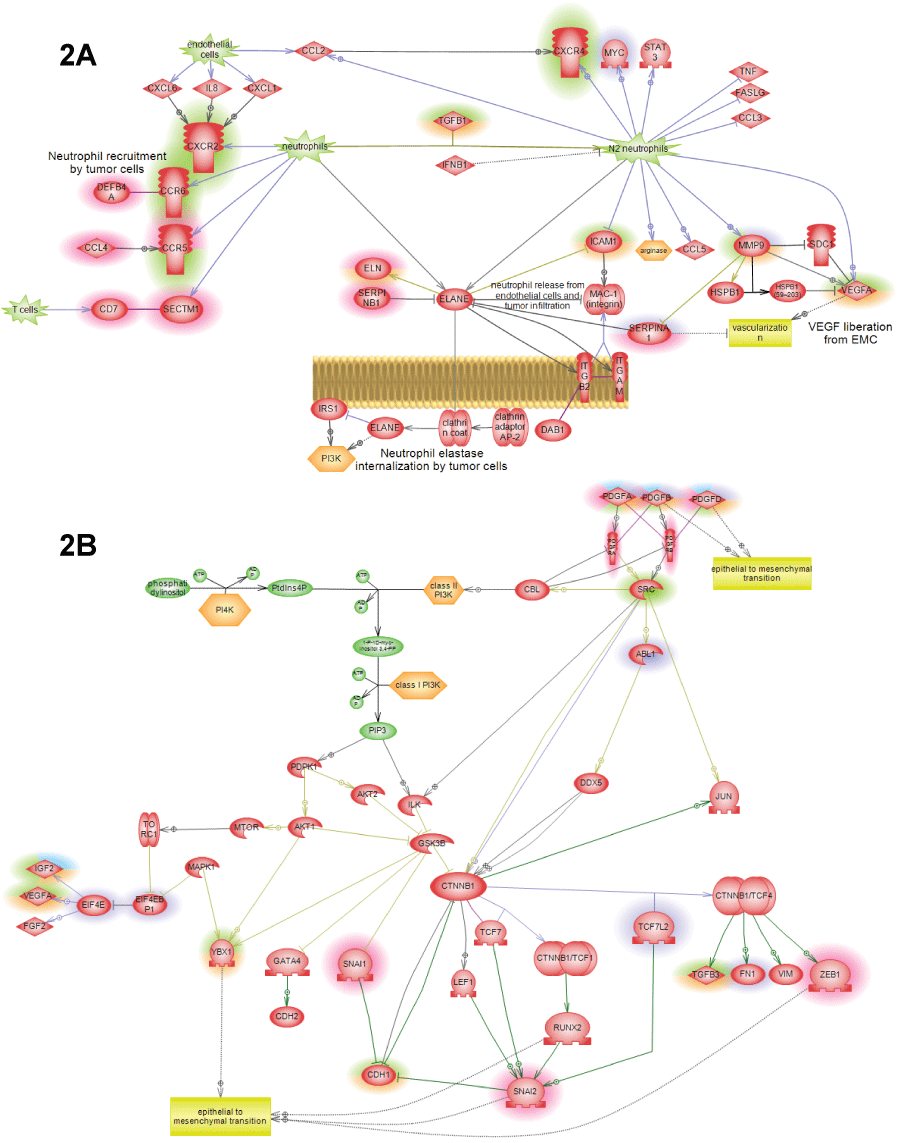
.
Figure 2: Additional pathways showing SNEA expression regulators identified in cancer patients by highlight. Different highlight colors correspond to different patients used in this study: orange – first lung cancer patient; green – second lung cancer patient; red – liver cancer patient; figure legend is shown on figure 1A. All pathways were enriched with major expression regulators identified by SNEA with p-value smaller than 0.05 according to Fisher exact test.
A) N1->N2 neutrophil polarization pathway showing how tumor causes re-differentiation on invading neutrophils into N2 phenotype that can promote tumor growth through the release of angiogenic factors and induction of migration; B) PDGF->EMT pathway showing how epithelial-to-mesenchymal transition is caused by the family of PDGF hormones through the induction of the standard EMT transcriptions factors: SNAIL, ZEB and catenin-neta.
View Figure 2A&B
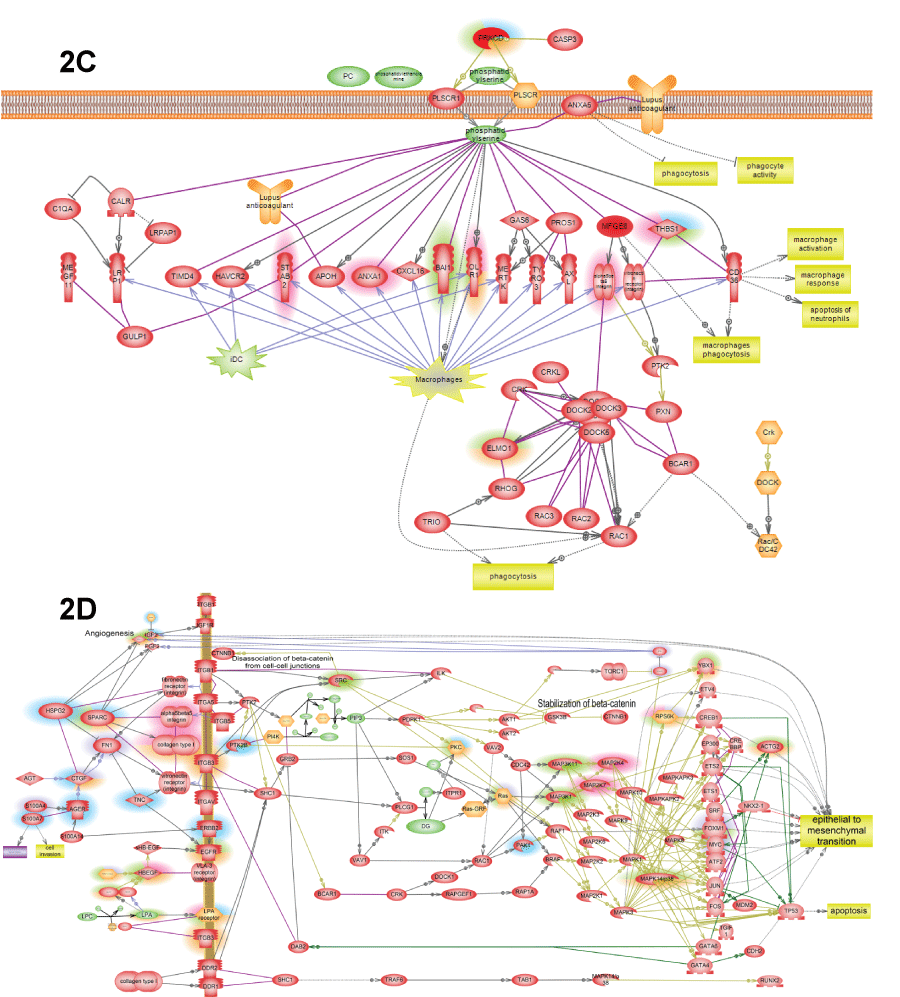
.
Figure 2: C) Phosphatidylserine is the major "eat me" signals of apoptotic cells inducing macrophage phagocytosis of apoptotic debris; D) ECM->EMT pathway shows how epithelial-to-mesenchymal transition is induced by several extracellular matrix (ECM) proteins that induce integrin outside-in signaling that lead both to activation of proliferation and invasive phenotype due to activation of FoxM1 and YBX1.
View Figure 2C&D
Table 4 lists most common SNEA regulators found among three patients. In summary, we found that angiogenesis in patients was driven by hypoxia via activation of HIF1 transcription factor, by thrombosis via activation of thrombin (F2), and by oxidized LDL. EGFR activation was due to macrophages activation releasing of oncostatin and HB-EGF. Macrophages were activated mainly by apoptotic debris clearance and by oxidized LDL. Apoptotic debris clearance is also known to induce HB-EGF conversion into active form via lysophosphatidic acid accumulation [16]. Tumor invasiveness was driven by epithelial-to-mesenchymal transition (EMT) promoted by hormones PDGF, IGF, TGFB; extracellular matrix proteins CTGF and SPARC; and transcription factor KLF5. Tumor proliferation was sustained through activation of EGFR. Observed activation of components of dREAM complex (LIN9, MYBL2, E2F3) responsible for transition between quiescent and proliferative states probably reflects tumor heterogeneity with part of the tumor being quiescent and not going through angiogenic switch [17].
![]()
Table 2: Representative cancer pathways enriched by top SNEA regulators found in patient tumors. Two metastatic tumors in lung from form the patient with metastatic colon cancer were analyzed. Receptor signaling pathways were considered activated only if at least one of its hormones or receptors was among SNEA regulators in patient. Such key regulators are shown in bold font in the last column. First column shows the tumor type for which the pathway was built based on SNEA regulators profile. All pathways were enriched with major expression regulators identified by SNEA with p-value smaller than 0.05 according to Fisher exact test.
View Table 2
![]()
Table 3: Cancer pathways activated in all three patients. All pathways were enriched with major expression regulators identified by SNEA with p-value smaller than 0.05 according to Fisher exact test.
View Table 3
Treatment selection for patients using pathway analysis
Figure 3 shows several examples of finding the drugs for a patient using pathway analysis in combination with ChemEffect knowledgebase available for Pathway Studio. ChemEffect contains regulatory effects for drugs found in scientific literature [15]. The main criteria for drug selection was that the drug had to be approved by FDA for cancer treatment or at very least allowed for cancer clinical trial. We then searched for drugs that had efficacy against pathways found to be activated in patient tumor according to SNEA analysis. If pathway did not have any druggable components we resorted to FDA-approved drugs that showed efficacy toward cell process that was found to be activated in patient tumor, e.g. epithelial-to-mesenchymal transition (EMT) in figure 3B. If several drugs satisfied above criteria we selected a drug which efficacy against the same type of tumor as in patient had the most support in the literature, i.e., had the biggest number of articles and clinical trials describing drug efficacy towards the appropriate cancer.
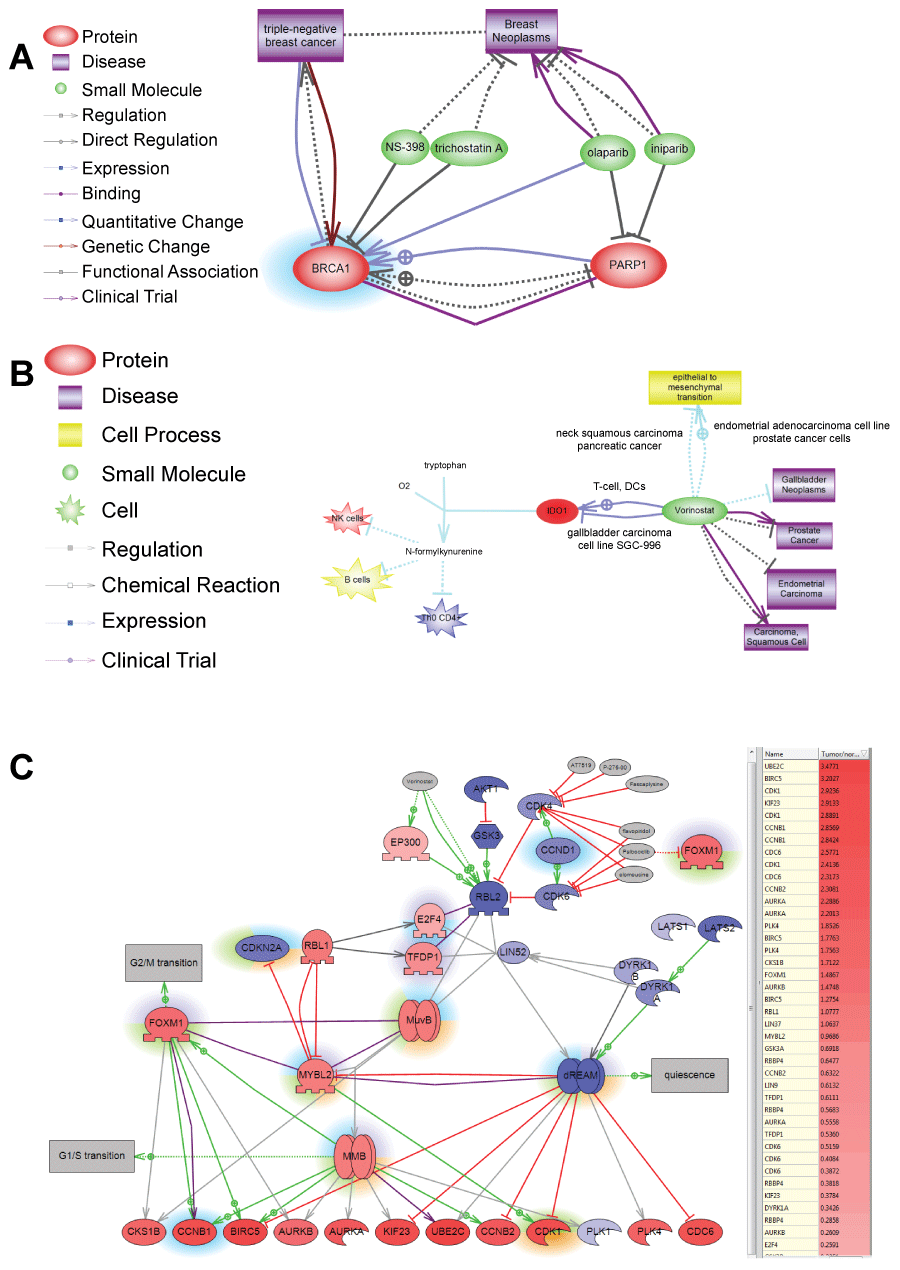
.
Figure 3: A) Shows example of selecting using ChemEffect knowledgebase in Pathway Studio. BRCA1 protein was found to be a major expression regulator in patient with breast cancer metastasis in the lung. While NS-398 and trichostatin A were shown to inhibit BRCA1 [52,53] there were not FDA approved for cancer treatment. Olaparib and iniparib, however, were in several clinical trials for breast cancer as indicated by http://clinicaltrial.gov. Both drugs inhibit PARP1– the direct upstream activator of BRCA1 in breast cancer; B) Shows selection of Vorinostat for patient with liver cancer based on the efficacy towards epithelial-mesenchymal transition. Pathway analysis of SNEA regulators found that tumor had hypoxia-induced EMT. Unfortunately, EMT is driven by transcription factors such as ZEB1/2, SNAIL1/2, KLF5 that are not druggable. Vorinostat, however, is the only FDA-approved drug that is known to inhibit epithelial-mesenchymal transition and was effective against several types of cancer in clinical trials. Figure shows the mechanism for Vorinostat action in gallbladder cell line by breaking tumor immune-tolerance through inhibition of indoleamine 2,3-dioxygenase (IDO1) [54]; C) Shows selection of drugs inhibiting DREAM complex in patient with lung cancer. DREAM transcription factor complex governs cell transition from quiescent to proliferative state. The gene expression of its components is shown by color of protein nodes: red – protein expression is up-regulated in the tumor compared with syngenic control of healthy lung; blue - protein expression is down-regulated in the tumor. Different highlights around the proteins show SNEA regulators identified in different cancer patients. SNEA regulators from the lung cancer patient whose expression profile is shown on the pathwayis highlighted in green. Because principal components of the DREAM complex are not druggable pathway analysis found drugs inhibiting its upstream regulator – CDK4 kinase. One of these inhibitors Palbociclib (Ibrance) is also known to indirectly inhibit FoxM1 transcription factor important not only for cell proliferation but also for invasive tumor phenotype through activating epithelial-to-mesenchymal transition.
View Figure 3
![]()
Table 4: List of most common SNEA regulators identified in five tumors from three cancer patients.
View Table 4
Discussion
Novel approach for selection of personalized cancer therapy
We describe the approach for analysis of cancer tumor that enables rational therapy design based on molecular mechanism responsible for tumor malignancy in a patient. Our approach consists of identification of major regulators responsible for differential gene expression in patient tumor and subsequent identification of cancer hallmark pathways enriched with the regulators. To enable the workflow we constructed 49 pathways in Pathway Studio depicting various mechanisms of cancer development reported in scientific literature. While the underlying technology can be and should be optimized further our goal was to achieve validation of the approach in clinical settings. The drugs rationally selected based on pathway analysis prolonged patient survival beyond Overall Survival estimates based on standard of care treatment. Our study demonstrates that the analysis of individual tumor sample compared to multiple normal controls is sufficient to produce biologically relevant results that can be further used as basis for clinical decision to select personalized therapeutic intervention. The biological relevance of individual patient tumor analysis is confirmed by finding canonical text-book pathways activated in the tumor.
We believe that further improvements in technology will enable even more precise treatments for cancer patients. One major improvement can come from the manual curation of the Expression regulatory relations used by SNEA to calculate major regulators. The curation can not only reduce the number of false positives but also annotate relations with correct effect sign (positive or negative) and with tissue-specific information. The curation of effect sign will allow calculation of the correct activation score for each SNEA regulator based on the concordance between the directions of expression change of each target and effect of the regulation event (i.e., transcription activation or repression). Such activation score can be used for Gene Set Enrichment analysis (GSEA) of cancer pathways [9,18] instead of overlap Fisher’s Exact test that was used in this article. Annotation with tissue specificity should also improve accuracy of SNEA p-value calculation that is used to determine regulator significance. Our cancer pathway collection can be also expanded since it does not yet contain pathway for all cancer hallmarks [13]. We have only built pathways containing SNEA regulators in order to interpret microarray data from our patients.
The fact that all our patients outlived the life expectancy from the current standard of care suggests that individualized therapy selected based on molecular profile of the tumor can be better alternative to one drug fits all patients model suggested by the standard of care approach. We also argue that selection of patients for clinical trials based on the molecular profile of the tumor using our approach can significantly improve chances for the trial success.
Cellular mechanisms found in patients with advanced cancers
Our approach identifies cell processes driving cancer progression in a patient. These processes can be used for drug selection. All three patients analyzed for this article had advanced cancers characterized by metastasis and rapid tumor growth according to clinical parameters. Consequently, we have identified processes activated in these patients that confer advanced tumorigenesis. Several pathways were built by translation of data obtained from in vitro studies for patient data. Identification of SNEA regulators from these translational pathways in patient tumors further confirmed that these in vitro findings were applicable in clinics. For example, pathway "EGFR activation by apoptotic clearance" was built by combining the established role of lysophosphatidic acid (LPA) in the activation of EGFR signaling in cancer [19,20] with very recent in vitro studies showing that tumor-associated macrophages is the source of HB-EGF, which is EGFR ligand that is activated by LPA [21]. We hypothesize that tumor-associated macrophages are activated by apoptotic debris that is in abundance inside the tumor. Lysophosphatidylcholine (LPC) is the major "Find-me" signal attracting macrophages to apoptotic debris [22,23] and at the same time is precursor of LPA. Thus, dying tumor cells activate EGFR pathway through macrophage activation to sustain tumor growth. "EGFR activation by apoptotic clearance" pathway resembles wound healing mechanism that was adapted for tumor growth.
We observed thrombin activation in all three patients (Table 3 and Table 4). This further supports a role of platelet-induced coagulation in tumor progression [24-26]. Thrombin can promote several cancer hallmarks. It sustains tumor growth via activation of oncogenic AP-1transription factor. Thrombin activates HIF1A transcription factor [27] which mimics hypoxic state and leads to angiogenesis by activating endothelial cells and to macrophage polarization enabling tumor to avoid immune destruction [28]. Thrombin also activates ROCK kinase causing cytoskeleton reorganization. Cytoskeleton reorganization in endothelial cells promotes their migration and therefore further promotes angiogenesis and vascular permeability; on the other hand activation of cytoskeleton reorganization in tumor cells promotes its invasiveness [29,30].
We developed "TGF-beta loop" pathway that depicts mechanism of epithelial-to-mesenchymal transition induced by TGF-β mediates. TGF-β is potent autocrine inducer of epithelial-to-mesenchymal transition in the presence of growth factors [30]. While the autocrine secretion of TGF-β in tumors was described previously [31] as TGF-β->SNAIL->SMAD3/4-> TGF-β loop [32-36] we were able to infer the existence of multiple secondary loops activating latent TGF-β activation from existing literature. These secondary loops are mediated by RUNX transcription factors [37] and their dimerization with ETS1/2 on target gene promoters [38-40]. RUNX induces expression of TGF-β receptors [38], while ETS1 activates expression of integrin-beta 6 [41], CD44 [42] and FLT1 [43]. AlphaVbeta6 integrin complex binds and activates latent TGF-β [44], CD44 binds TGF-β receptors [45] and activates latent TGF-β [46], FLT1 mediates survival in post-EMT cancer cells [47]. Both RUNX and ETS1 activate expression of MMP9 [48,49], which cleaves and activates latent TGF-β [50,51]. RUNX transcription factor was shown to be activated in metastatic breast cancer [48] and in thyroid carcinoma [49].
We found that the similarity between patients was higher on pathway level than on the level of individual expression activators. Figure 2 depicts pathways identified in different patients with their respective expression activators highlighted by one color corresponding to each patient. Thus, even if patients had different set of expression regulators they still pointed to the same pathway suggesting that the same pathway can be activated in individual tumors albeit different mechanisms. Similarity of tumors on pathway level may be explained by the fact that all patients had metastatic cancer. Because we selected the same treatments for patients with similar activated pathways our finding also suggests that patient with similar activated pathways can have similar clinical outcome despite the difference in the underlying molecular mechanism for pathway activation in their tumors.
Drug availability to target patient pathways
The most logical choice of anti-cancer therapy is a drug inhibiting major expression regulators activated in patient tumor. Our experience with three patients revealed that many regulators, especially transcription factors, do not have either direct or indirect FDA approved inhibitors, while many existing FDA-approved inhibitors found in Pathway Studio are not approved for cancer treatment by FDA. Therefore for most patients we resorted to experimental drugs that have shown some efficacy against appropriate type of cancer and also could inhibit the cell process indicated by pathway analysis (Table 3). Noteworthy, in cases when FDA-approved drugs were not available for SNEA regulators we always were able to find plant extracts with reported inhibitory properties.
References
-
MAQC Consortium, Shi L, Reid LH, Jones WD, Shippy R, et al. (2006) The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol 24: 1151-1161.
-
Chen JJ, Hsueh HM, Delongchamp RR, Lin CJ, Tsai CA (2007) Reproducibility of microarray data: a further analysis of microarray quality control (MAQC) data. BMC Bioinformatics 8: 412.
-
Chibon F (2013) Cancer gene expression signatures - the rise and fall? Eur J Cancer 49: 2000-2009.
-
Michiels S, Kramar A, Koscielny S (2011) Multidimensionality of microarrays: statistical challenges and (im)possible solutions. Mol Oncol 5: 190-196.
-
Hua J, Xiong Z, Lowey J, Suh E, Dougherty ER (2005) Optimal number of features as a function of sample size for various classification rules. Bioinformatics 21: 1509-1515.
-
Valletti A, Gigante M, Palumbo O, Carella M, Divella C, et al. (2013) Genome-wide analysis of differentially expressed genes and splicing isoforms in clear cell renal cell carcinoma. PLoS One 8: e78452.
-
Chan SK, Griffith OL, Tai IT, Jones SJ (2008) Meta-analysis of colorectal cancer gene expression profiling studies identifies consistently reported candidate biomarkers. Cancer Epidemiol Biomarkers Prev 17: 543-552.
-
Pyatnitskiy M, Mazo I, Shkrob M, Schwartz E, Kotelnikova E (2014) Clustering gene expression regulators: new approach to disease subtyping. PLoS One 9: e84955.
-
Sivachenko AY, Yuryev A, Daraselia N, Mazo I (2007) Molecular networks in microarray analysis. J Bioinform Comput Biol 5: 429-456.
-
Catlett NL, Bargnesi AJ, Ungerer S, Seagaran T, Ladd W, et al. (2013) Reverse causal reasoning: applying qualitative causal knowledge to the interpretation of high-throughput data. BMC Bioinformatics 14: 340.
-
Zhang L, Zhang J, Yang G, Wu D, Jiang L, et al. (2013) Investigating the concordance of Gene Ontology terms reveals the intra- and inter-platform reproducibility of enrichment analysis. BMC Bioinformatics 14: 143.
-
Abatangelo L, Maglietta R, Distaso A, D'Addabbo A, Creanza TM, et al. (2009) Comparative study of gene set enrichment methods. BMC Bioinformatics 10: 275.
-
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646-674.
-
Lorenzi PL, Claerhout S, Mills GB, Weinstein JN (2014) A curated census of autophagy-modulating proteins and small molecules: candidate targets for cancer therapy. Autophagy 10: 1316-1326.
-
Yuryev A, Kotelnikova E, Daraselia N (2009) Ariadne's ChemEffect and Pathway Studio knowledge base. Expert Opin Drug Discov 4: 1307-1318.
-
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545-15550.
-
Tanaka Y, Miyamoto S, Suzuki SO, Oki E, Yagi H, et al. (2005) Clinical significance of heparin-binding epidermal growth factor-like growth factor and a disintegrin and metalloprotease 17 expression in human ovarian cancer. Clin Cancer Res 11: 4783-4792.
-
Wang SH, Lin SY (2013) Tumor dormancy: potential therapeutic target in tumor recurrence and metastasis prevention. Exp Hematol Oncol 2: 29.
-
Braun AH, Coffey RJ (2005) Lysophosphatidic acid, a disintegrin and metalloprotease-17 and heparin-binding epidermal growth factor-like growth factor in ovarian cancer: the first word, not the last. Clin Cancer Res 11: 4639-4643.
-
Wu J, Cunnick JM (2002) Trans-regulation of epidermal growth factor receptor by lysophosphatidic acid and G protein-coupled receptors. Biochim Biophys Acta 1582: 100-106.
-
Vlaicu P, Mertins P, Mayr T, Widschwendter P, Ataseven B, et al. (2013) Monocytes/macrophages support mammary tumor invasivity by co-secreting lineage-specific EGFR ligands and a STAT3 activator.BMC Cancer 13: 197.
-
Hochreiter-Hufford A, Ravichandran KS (2013) Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol 5: a008748.
-
Lauber K, Ernst A, Orth M, Herrmann M, Belka C (2012) Dying cell clearance and its impact on the outcome of tumor radiotherapy. Front Oncol 2: 116.
-
Goubran HA, Stakiw J, Radosevic M, Burnouf T (2014) Platelet-cancer interactions. Semin Thromb Hemost 40: 296-305.
-
Goubran HA, Burnouf T, Radosevic M, El-Ekiaby M (2013) The platelet-cancer loop. Eur J Intern Med 24: 393-400.
-
Goubran HA, Burnouf T (2012) Platelets, coagulation and cancer: Multifaceted interactions. American Medical Journal 3: 130-140.
-
Brahimi-Horn MC, Pouysségur J (2005) The hypoxia-inducible factor and tumor progression along the angiogenic pathway. Int Rev Cytol 242: 157-213.
-
Escribese MM, Casas M, Corbí AL (2012) Influence of low oxygen tensions on macrophage polarization. Immunobiology 217: 1233-1240.
-
Shi J, Wei L (2013) Rho kinases in cardiovascular physiology and pathophysiology: the effect of fasudil. J Cardiovasc Pharmacol 62: 341-354.
-
Huber MA, Kraut N, Beug H (2005) Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol 17: 548-558.
-
Moustakas A, Heldin CH (2012) Induction of epithelial-mesenchymal transition by transforming growth factor β. Semin Cancer Biol 22: 446-454.
-
Dave N, Guaita-Esteruelas S, Gutarra S, Frias À, Beltran M, et al. (2011) Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem 286: 12024–12032.
-
Medici D, Hay ED, Olsen BR (2008) Slug promote epithelial–mesenchymal transition through ß-catenin-T-cell factor-4-dependent expression of transforming growth factor-β3. Molecular Biology of the Cell 19: 4875–4887.
-
Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, et al. (2010) Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proceedings of the National Academy of Sciences of the United States of America, 107: 15449-15454.
-
Thuault S, Tan EJ, Peinado H, Cano A, Heldin CH, et al. (2008) HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J Biol Chem 283: 33437-33446.
-
Chimge NO, Baniwal SK, Little GH, Chen YB, Kahn M, et al. (2011) Regulation of breast cancer metastasis by Runx2 and estrogen signaling: the role of SNAI2. Breast Cancer Res 13: R127.
-
Sancisi V, Gandolfi G, Ragazzi M, Nicoli D, Tamagnini I, et al. (2013) Cadherin 6 is a new RUNX2 target in TGF-β signalling pathway. PLoS One 8: e75489.
-
Erman B, Cortes M, Nikolajczyk BS, Speck NA, Sen R (1998) ETS-core binding factor: a common composite motif in antigen receptor gene enhancers. Mol Cell Biol 18: 1322-1330.
-
Kim WY, Sieweke M, Ogawa E, Wee HJ, Englmeier U, et al. (1999) Mutual activation of Ets-1 and AML1 DNA binding by direct interaction of their autoinhibitory domains. EMBO J 18: 1609-1620.
-
Sun W, Graves BJ, Speck NA (1995) Transactivation of the Moloney murine leukemia virus and T-cell receptor beta-chain enhancers by cbf and ets requires intact binding sites for both proteins. J Virol 69: 4941-4949.
-
Bates RC, Bellovin DI, Brown C, Maynard E, Wu B, et al. (2005) Transcriptional activation of integrin beta6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J Clin Invest 115: 339-347.
-
Zhang W, Zhao J, Lee JF, Gartung A, Jawadi H, et al. (2013) ETS-1-mediated transcriptional up-regulation of CD44 is required for sphingosine-1-phosphate receptor subtype 3-stimulated chemotaxis. J Biol Chem 288: 32126-32137.
-
Wakiya K, Begue A, Stehelin D, Shibuya M (1996) A cAMP response element and an Ets motif are involved in the transcriptional regulation of flt-1 tyrosine kinase (vascular endothelial growth factor receptor 1) gene. J Biol Chem 48: 30823-30828.
-
Worthington JJ, Klementowicz JE, Travis MA (2011) TGFβ: a sleeping giant awoken by integrins. Trends Biochem Sci 36: 47-54.
-
Pardali K, Moustakas A (2007) Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta 1775: 21-62.
-
Bellon G, Martiny L, Robinet A (2004) Matrix metalloproteinases and matrikines in angiogenesis. Crit Rev Oncol Hematol 49: 203-220.
-
Bates RC, Goldsmith JD, Bachelder RE, Brown C, Shibuya M, et al. (2003) Flt-1-dependent survival characterizes the epithelial-mesenchymal transition of colonic organoids. Curr Biol 13: 1721-1727.
-
Pratap J, Javed A, Languino LR, van Wijnen AJ, Stein JL, et al. (2005) The Runx2 osteogenic transcription factor regulates matrix metalloproteinase 9 in bone metastatic cancer cells and controls cell invasion. Mol Cell Biol 25: 8581-8591.
-
Seth A, Watson DK (2005) ETS transcription factors and their emerging roles in human cancer. Eur J Cancer 41: 2462-2478.
-
Jenkins G (2008) The role of proteases in transforming growth factor-beta activation. Int J Biochem Cell Biol 40: 1068-1078.
-
Centrella M, McCarthy TL (2012) Estrogen receptor dependent gene expression by osteoblasts - direct, indirect, circumspect, and speculative effects. Steroids 77:174-184.
-
Gao XQ, Han JX, Huang HY, Song B, Zhu B, et al. (2005) Effect of NS398 on metastasis-associated gene expression in a human colon cancer cell line. World J Gastroenterol 11: 4337-4343.
-
Molli PR, Singh RR, Lee SW, Kumar R (1971) MTA1-mediated transcriptional repression of BRCA1 tumor suppressor gene. Oncogene 27: 1971-1980.
-
Zhang P, Jiang G, Gao J, Li L, Du J, et al. (2013) SAHA down-regulates the expression of indoleamine 2,3-dioxygenase via inhibition of the JAK/STAT1 signaling pathway in gallbladder carcinoma cells. Oncol Rep 29:269-275.
